Синдром Марфана — Википедия
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Наследственное заболевание.
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах. Мужчины и женщины страдают с одинаковой частотой[3].
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения[4]. В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном, который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией.[6]
Американский генетик Виктор Маккьюсик открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани.[7]
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.[8]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).
Диагностика синдрома Марфана (СМ) сегодня основана на Гентских критериях (DeРаере A. et al.,1996) и пересмотра Гентских критериев в 2010 году (J.Med.Genet.2010; 476-485). В основу алгоритма диагностики положено выделение больших и малых критериев, характеризующих выраженность изменений соединительной ткани в различных органах и системах.
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Синдром Марфана (неопр.) (недоступная ссылка). Первый Московский Государственный Университет имени И.М. Сечнова (19 октября 2007). — Течение и прогноз. Дата обращения 23 августа 2012. Архивировано 14 августа 2011 года.
- ↑ Marfan Syndrome (англ.). NORD (National Organization for Rare Disorders). Дата обращения 23 сентября 2019.
- ↑ Williams E. Rare Cases, with Practical Remarks Архивная копия от 20 декабря 2016 на Wayback Machine // Transactions of the American Ophthalmological Societ. — 1875. — Vol. 2. — PP. 291—301. — PMC 1361735
- ↑ Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- ↑ Hecht F., Beals R. K. «New» syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. // Pediatrics. — 1972 Apr. — Vol. 49(4). — PP. 574—579. — PMID 4552107
- ↑ McKusick V. A.
- ↑ Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- ↑ Goldman’s Cecil Medicine 978-1-4557-1167-3,
- Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
На русском языке[править | править код]
На английском языке[править | править код]
Синдром Марфана: причины, симптомы, лечение, прогноз

Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.

мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей.

Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
-
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении; - выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
 аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Классификация
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.

Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.

Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,
- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.

Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
- страбизм,
- катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
 страбизм,
страбизм,Прочие органы
Поражение нервной системы:
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
Поражение бронхопульмональной системы:
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.
 У больных пневмотораксом воздух скапливается в плевральной полости, а легкие сжимаются. Их дыхание становится частым и поверхностным, в груди болит, кожные покровы сначала бледнеют, а затем приобретают синюшный оттенок.
У больных пневмотораксом воздух скапливается в плевральной полости, а легкие сжимаются. Их дыхание становится частым и поверхностным, в груди болит, кожные покровы сначала бледнеют, а затем приобретают синюшный оттенок.
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методы диагностики

проявления синдрома Марфана в младшем возрасте
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Методы лечения

Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Медикаментозное лечение:
- β-адреноблокаторы – «Пропранолол», «Атенолол»;
- блокаторы кальциевых каналов – «Нифедипин», «Верапамил»;
- ингибиторы АПФ – «Каптоприл», «Лизиноприл»;
- коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
- метаболики – «Рибоксин», «Милдронат»;
- поливитаминные комплексы;
- антибиотики для профилактики инфекционного эндокардита;
- антикоагулянты для профилактики тромбоза;
- антиоксиданты и антигипоксанты – «Коэнзим Q10», «Элькар»;
- ноотропные препараты – «Пирацетам», «Винпоцетин».
 коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;
коллагеннормализующая терапия – «Алфлутоп», «Румалон», «Структум»;При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
Хирургическое лечение:
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Клинические рекомендации специалистов своим пациентам:
- ведение здорового образа жизни,
- ограничение тяжелого труда и физического перенапряжения,
- отказ от спортивных игр повышенной активности,
- регулярное посещение специалистов в области кардиологии, офтальмологии, ортопедии, неврологии,
- нахождение больных под постоянным наблюдением и контролем врачей,
- периодическое прохождение диагностического обследования.
Прогноз и профилактические мероприятия

Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. При наличии серьезных нарушений со стороны этих органов длительность жизни пациентов ограничивается 40-50 годами, и повышается риск внезапной смерти. С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность.
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Видео: синдром Марфана в программе “Жить здорово!”
Видео: ток-шоу о синдроме Марфана
Синдром Марфана; симптомы, лечение, диагностика, профилактика
У человека много генетических заболеваний, одним из таких проявляется синдром Марфана, который еще называется «паучьи пальцы». Основные симптомы синдрома Марфана связаны с нарушениями со стороны соединительной ткани. У больных людей нарушается производство белков в организме, из них потом синтезируется соединительная ткань. Болезнь прогрессирует, в результате чего у людей развиваются тяжелые нарушения, приводящие к инвалидности. Заболевание носит генетический характер, передается по наследству.
Немного истории
Первым описал болезнь, которая впоследствии получила имя Марфана врач Вильямсон в 1876 году. А название болезнь получила в честь педиатра из Франции по имени Марфан, который по прошествии двух десятилетий наблюдал ребенка, страдающего этой патологией. Известно, что болели известные люди, к примеру, Лесли Хорнби, которая стала прототипом эталона модельной индустрии. Также известные А. Линкольн, президент США, скрипач Паганини, как оказалось, имели синдром Марфана.
Потом по мере изучения стало известно: болезнь носит генетический характер, передается по наследству. Достаточно быть носителем синдрома патологического гена Марфана одному из родителей, чтобы потомство было больным. При отсутствии адекватного лечения синдрома Марфана, в том числе оперативного, прогноз неблагоприятный. Если не предпринимать мер, человек может не дожить до тридцати лет.
Симптоматика болезни
Симптомы сопровождаются поражением многих внутренних органов, поскольку соединительная ткань есть везде. Наиболее часто люди страдают нарушениями со стороны опорно-двигательного аппарата, сердечно-сосудистой, дыхательной, центральной нервной системы, нарушается зрение.
Симптомы проявляются астеническим типом телосложения: при высоком росте, свыше 180 сантиметров, очень маленькая масса тела, не больше 50 килограммов. Помимо этого, у людей очень длинные пальцы рук, ног, которые называют «паучьими пальцами». Деформируется ось позвоночника в виде кифосколиоза, грудная клетка становится килевидной, присоединяется плоскостопие. Лицо становится узким, небе высокое, готическое. Для людей с синдромом Марфана это наиболее распространенные симптомы, могут встречаться в отдельности или все вместе.
Синдром Марфана имеет типичное проявление: антимонголоидный разрез глаз, нос крупный, уши чрезмерно большие, расположены низко, лицо напоминает птицу. У новорожденного ребенка синдром Марфана проявляется только аномальным ростом, слишком длинными пальцами. Остальные симптомы проявляются на поздних сроках, как правило, на протяжении первых пяти или семи лет жизни.
Люди, страдающие заболеванием, имеют патологию со стороны сердца, сосудов. Развивается пролапс в области митрального клапана, аневризма (выпячивание) самого крупного сосуда – аорты. Вышеуказанные нарушения выявляются в течение первого или второго года жизни ребенка. Аорта увеличивается в диаметре, постепенно она достигает критического диаметра (более 6 см).
С возрастом, примерно от 16 до 45 лет, проявляется в полной мере. Самым грозным осложнением (оно способно возникнуть в любой момент) считается постоянное расслоение стенок аорты, при синдроме Марфана оно прогрессирует, постепенно распространяясь по всей длине сосуда, в отходящие ветви. Результатом является летальный исход: аорта может разорваться в любой момент, став причиной массивного кровотечения.
Часто люди, имеющие синдром Марфана, страдают патологией легких, бронхов. Причиной является деформированная грудная клетка из-за изменений соединительной ткани, которая входит в состав ткани дыхательной системы. Основные симптомы дополняет нарушение дыхания в качестве спонтанного пневмоторакса, не редкость такое состояние, как легочная эмфизема, инфаркт легкого встречается от 10% до 75 % при синдроме Марфана. Описаны ситуации врожденной аномалии легочной доли, булезной эмфиземы поликистоз, двухсторонних бронхоэктазов.
Со стороны зрения у людей, имеющих синдром Марфана, развивается смещение хрусталика глаза. Причиной подобного состояния является слабость связки, которая его удерживает. В дополнение у человека развивается близорукость или дальнозоркость, выраженная в сильной степени. Смещение хрусталика подтверждается в срок от первого до пятого года жизни, порой родители ничего не знают до 7 лет, все становится известно, когда ребенок идет в школу. Вторичным состоянием считается глаукома, катаракта, может отслоиться сетчатка. Изменения наблюдаются от 15 до 40 лет, ясно указывают на наличие патологии Марфана.
Человек, больной синдромом Марфана, имеет сниженное умственное развитие. Ребенок плаксив, раздражителен, имеет завышенную самооценку. Снижена устойчивость к физической нагрузке, в результате ее наступает быстрое переутомление мышц, боль. Во время или после психоэмоциональных нагрузок возникает головная боль по типу мигрени. Ребенок имеет слаборазвитые мышцы, для их укрепления можно использовать профилактор Евминова.
Постановка диагноза
Точная, своевременная диагностика синдрома Марфана способна вызвать определенные трудности, для врача это просто. Установить истину позволяет анализ родословной, исследование генов. Помимо симптомов, изучается физическое, нервно-психическое развитие. Физическое состояние изучается при помощи шкалы Стюарта.
Поставить диагноз «синдром Марфана» можно, если есть один из признаков, к примеру, вывих или частичное смещение хрусталика, аневризма аорты, аномально длинные, похожие на лапы паука, пальцы (арахнодактилия), деформация любой части грудной клетки, искривление позвоночника.
Но этого маловато, чтобы диагностировать болезнь Марфана, потребуется еще два фактора: близорукость, пролапс в области митрального клапана, подвижность суставов умеренной степени выраженности, аномально высокий рост, плоскостопие, «растяжки» на коже, пневмоторакс.
В 90% случаев диагностировать синдром Марфана удается с первого раза, при остальных 10% могут возникнуть определенные трудности. В подобной ситуации обследованию подлежит максимальное число родственников. В обязательном порядке требуется консультация генетика, офтальмологическое, кардиологическое обследование, исследование эхокардиографии. Дополнить представление врача о синдроме Марфана может рентген, измеряются индексы длины пальцев. Они вычисляются на основании рентгенологического снимка, если есть синдром Марфана, индекс будет удлинен. Определяется он на соотношении длины к толщине второй пястной кости.
Выявить характер, тяжесть заболевания сердца позволяет установить эхокардиография, ЭКГ, холтеровское мониторирование. Оценить состояние легких, бронхов позволяет определение функции внешнего дыхания. При заболевании в большинстве обнаруживаются характерные нарушения. Характерные изменения наблюдаются и в анализе мочи. Дополнительно с целью диагностики составляется генеалогическое дерево, позволяющее определить взаимосвязь заболевания, как оно передается. Составляется оно по третье поколение, но чем больше известно информации о родственниках более ранних поколений, точнее будет диагностика.
Отсеять арахнодактилию
Чтобы начать лечение синдрома Марфана или арахнодактилии необходимо отсеять другие заболевания, имеющие сходную клиническую картину. Так, астеническое телосложение встречается при болезни Марфана, Билса, Стиклера, но этого симптома нет у болезни Вейла-Мерчезани. Арахнодактилия присутствует при всех заболеваниях, кроме последнего, есть аутосомно-доминантный тип передачи. Чаще всего при болезни Марфана встречается патология аорты, смещение хрусталика.
Лечение арахнодактилии
Лечить синдром Марфана можно только комплексно с использованием многих препаратов. В некоторых ситуациях используется профилактор Евминова. Применяют препараты для восстановления деятельности сердечно-сосудистой системы, стимулирующие мозг, енерготропные препараты, антиоксиданты. Все препараты применяются строго по определенной схеме, которая специально разработана для этой патологии.
Дополняет медикаментозное лечение физиотерапевтическое, которое включает в себя магнит. Курс состоит из десяти сеансов, назначаемых три раза на протяжении года. Показан электросон, курс которого состоит из десяти посещений два раза в течение года. Для лечебной физкультуры можно использовать профилактор Евминова, гимнастика выполняется на протяжении двух недель, четыре раза на протяжении года. Последняя методика показана при патологии костей, суставов, для поддержания сердца применяется санаторно-курортное восстановление на протяжении 24 дней в течение календарного года. Дополнить лечение арахнодактилии может профилактор Евминова.
В дополнение проводится операция, пластика деформации грудной клетки, оперативно устраняется аневризма любой части аорты с последующей пластикой. Подвывих, полное смещение хрусталика подлежит оперативному лечению. Примерно два раза на протяжении года показана санация всех источников инфекции, в частности, зубов. При правильно назначенном лечении синдрома, если использовать профилактор Евминова, до 80% наблюдается стабилизация состояния.
Лечение, его эффективность при арахнодактилии оценивается по объективным лабораторным показателям. У малыша повышается выносливость к физическим нагрузкам, постепенно увеличивается мышечная масса. Стабилизируется диаметр аорты, в этом помогает эхокардиография. Приходит в норму функция внешнего дыхания, улучшается мелкая моторика, повышается эмоциональный тонус. Восстанавливается память, ребенок концентрирует внимание, успеваемость в школе становится выше. В лабораторных анализах при синдроме арахнодактилии или Марфана снижается уровень молочной, пировиноградной кислоты.
Режим труда, отдыха при синдроме арахнодактилии
Во время лечения болезни Марфана потребуется соблюдать некоторые рекомендации, они касаются труда, отдыха, восстановления. Физкультура, гимнастика, ЛФК при патологии Марфана проводятся по облегченной схеме в специальной группе, дополнительно можно использовать профилактор Евминова. Запрещено заниматься спортом в секциях, участвовать в соревнованиях, трудиться в сельском хозяйстве, ходить на продолжительные дистанции, особенно пересекать горные местности. Запрещено переносить, поднимать тяжести, максимальный вес составляет не больше 3 килограммов.
В будущем противопоказана работа, связанная с профессиональными вредностями. Такими считается контакт с химикатами, лакокрасочными материалами, труд при высокой температуре, а также воздействие радиации. Категорически нельзя выбирать профессии, связанные с вибрацией, когда требуется высокая острота зрения, противопоказаны физические, эмоциональные нагрузки.
При подборе места постоянного проживания больному синдромом Марфана противопоказан жаркий климат, зона повышенного уровня радиационного фона. Беременной, страдающей синдромом Марфана, не реже, чем раз в два месяца, показано проходить эхокардиографию. Когда диаметр аорты превышает 45 мм необходимо решать вопрос чем скорее, тем лучше о целесообразности, важности продолжения беременности. Есть угроза не только для плода, но и матери при патологии Марфана или синдроме арахнодактилии.
Показаны тренировки, где используется профилактор Евминова. Роды беременной, страдающей синдромом Марфана, проводятся только оперативным путем в специальных родильных отделениях.
Профилактика синдрома арахнодактилии
Перед тем как вступать в брак, оба супруга проходят медицинское обследование, обязательную консультацию у врача — генетика. Врач может спрогнозировать риск развития патологии у супругов. Дополнительно применяется пренатальное определение этого синдрома.
Профилактор
При заболевании для выполнения гимнастики, разгрузки позвоночника может применяться профилактор Евминова. Представляет он собой специальную доску, на вид она простая, но на самом деле позволяет укрепить спину. Длительность тренировок составляет от пятнадцати до двадцати минут в день. Если зона роста не закрыта, то профилактор Евминова позволяет сделать ее длиннее, а не только крепче, главное, заниматься регулярно.
Лечение профилактором
Восстановление, профилактика патологии спины проводится за счет комплекса гимнастики, которая выполняется в положении лежа, помогает в этом профилактор Евминова. Подбор комплекса для каждого человека проводится индивидуально в зависимости от физических возможностей организма человека. Внешне профилактор Евминова представлен деревянной панелью с подвижными ручками, расположенными на двух уровнях. Укрепляется конструкция к стене при помощи троса, устанавливается под углами.
Во время занятий на тренажере обеспечиваются два принципа, направленных на восстановление нормальной функции позвоночника. Во время первого производится контролируемое вытяжение позвоночного столба, вследствие чего обеспечивается его максимальная разгрузка. Степень вытяжения определяется за счет перемены угла наклона. Во время растяжения происходит расширение межпозвоночной щели, освобождаются сжатые нервные корешки, боль постепенно исчезает.
Суть второго принципа в том, что происходит не только тренировка, укрепление мускулатуры, поддерживающей позвоночник в горизонтальном уровне, это является нормальной анатомией. Тренировка, укрепление проводится при помощи специального комплекса упражнений, которые выполняются при минимальной амплитуде движения. За тренировку происходит укрепление маленьких мышц спины, которые собственно и образуют мышечный корсет.
Чем раньше начато лечение, тем больше процент стабилизации состояния. Болезнь носит врожденный генетический характер, вылечить ее полностью нельзя. Поможет врач — генетик, с ним придется проконсультироваться, особенно если кто-то из членов семьи (или предков) страдает болезнью Марфана.
что это такое, тип наследования, симптомы и возможность лечения
Синдром Марфана – редко встречающаяся генетическая патология, сопровождающаяся наличием дефектов в строении соединительной ткани. Поскольку данная структура входит в состав всех систем человеческого организма, то заболеванию свойственна неоднородность клинической картины. Она может включать в себя как симптомы поражения сердца, так и аномалии строения опорно-двигательного аппарата, а также неврологические расстройства. Специфических методов лечения и профилактики на сегодняшний день не разработано. Терапия направлена на предотвращение развития фатальных осложнений, а также на улучшение качества жизни пациентов с данным диагнозом.
Причины возникновения синдрома Марфана
Патология относится к врожденным заболеваниям, склонным к наследованию. Распространенность поражения невелика – регистрируется около одного случая на 20 тысяч человек. Гендерной и расовой зависимости медиками не установлено. Синдрому Марфана свойственен аутосомно-доминантный тип наследования. Риск формирования генетической мутации увеличивается по мере старения отца, что связано с накоплением ошибок в процессе деления клеток. В основе развития патологии лежит дефект участка хромосомы, отвечающего за синтез белка фибриллина. Внешних факторов, которые могли бы явиться причиной возникновения синдрома Марфана, на сегодняшний день не выявлено.

Влияние наследственных факторов
Патологии свойственно передаваться плоду от родителей. При этом подтверждение заболевания у матери или отца не гарантирует 100% вероятности выявления нарушения у ребенка. Проявление симптомов синдрома Марфана обусловлено наличием дефекта в строении белка FBN1. Эта структура отвечает за синтез элемента соединительной ткани, который обеспечивает ее эластичность и сопротивляемость физическим нагрузкам. Недостаточность фибриллина сопровождается неспособностью клеток к выполнению своих функций. Именно это нарушение приводит к возникновению характерных симптомов. Отмечается слабость связочного аппарата, а также крупных сосудов эластического типа. Наследственный вид патологии диагностируется в 70–75% случаев, имеет место и первичная проблема.
Стоит отметить, что синдром Марфана отличается разнообразием клинических признаков. Подобная разнородность связана с многочисленностью мутаций, обуславливающих возникновение болезни. В генетике описано около 1 тыс. вариаций дефектов FBN1.
Классификация и основные признаки
В медицине принято дифференцировать две формы болезни:
- Синдром Марфана выраженного типа сопровождается интенсивными клиническими проявлениями. У пациентов регистрируются отклонения в развитии 3-х различных систем организма, при этом в 2-х или 3-х они сопровождаются яркими симптомами.
- Стертая форма патологии характеризуется меньшими проявлениями. Тяжесть течения поражения индивидуальна в каждом отдельном случае и зависит от типа мутации соответствующего гена. Данному виду проблемы свойственно наличие слабо выраженных отклонений в 1-й – 2-х системах организма.
В ряде случаев диагноз звучит как «марфаноподобный синдром». Это означает положительную генетическую диагностику, но сглаженность клинической картины. Большинство врачей относят эту проблему к стертой форме проявления заболевания.
Синдром дифференцируют и по течению. Медики различают стабильный вариант нарушения и прогрессирующий. Первый сопряжен с врожденными аномалиями, которые легко поддаются диагностике и зачастую требуют радикального решения, вплоть до проведения операции. Если же интенсивность проявления симптомов нарастает, первоначально проводится поддерживающее лечение. Такая терапия направлена на стабилизацию состояния пациента.

Поражение сердца и сосудов
Соединительная ткань входит в состав всех систем человеческого организма. Наибольшее количество таких элементов в крупных артериях. Это необходимо для естественного тонуса сосудов, которые должны выдерживать значительное давление.
При наличии дефекта гена, ответственного за производство фибриллина, отмечается сбой в работе кардиальных структур. Распространенной проблемой считается потеря эластичности стенок аорты. Подобный каскад реакций обеспечивает расширение данной структуры, повышается риск развития аневризмы. При отсутствии лечения велика вероятность разрыва сосуда.
Соединительная ткань также входит в состав клапанов сердца, формируя тяжи, обеспечивающие работу кардиальных створок. Снижение плотности и эластичности этих соединений может сопровождаться формированием сердечной недостаточности. Больные страдают от непереносимости физических нагрузок, выраженной одышки. Слизистые приобретают бледный или синюшный цвет. Отмечается общая слабость, головокружение, перепады артериального давления.
Нарушения в работе опорно-двигательного аппарата
У пациентов с синдромом Марфана отмечаются особенности строения скелета. Подобные дефекты особенно заметны у детей и подростков в период активного развития костей и суставов. Синдром паучьих пальцев или арахнодактилия – характерный признак данной патологии. Скелет человека имеет вытянутые черты. Это связано с первостепенным поражением длинных костей опорно-двигательной системы. Больные зачастую страдают также от плоскостопия, деформаций грудины и искривления позвоночника. Лицо у пациентов вытянутое, запястья узкие. «Паучьи» пальцы тонкие, значительно выделяются по длине в соотношении с другими частями тела.
Проблемы со зрением
Дефицит соединительнотканных элементов в цинновой связке глаза сопровождается развитием офтальмологической симптоматики. Характерным является изменение расположения и формы хрусталика. Подобная проблема инициирует снижение остроты зрения. Опасное осложнение этого состояния – отслоение сетчатки. Патология по мере прогрессирования приводит к нетрудоспособности пациента. Она формируется вследствие полной потери зрения.
Изменения в легких
Симптомы со стороны респираторной системы у больных с синдромом Марфана регистрируются редко. Подобные нарушения связаны с изменением нормальной эластичности альвеол, которые могут растягиваться или спадаться. Среди врачей существует мнение, что данный дефект способен являться провоцирующим фактором в развитии рака легких. В редких случаях у пациентов отмечаются нарушения дыхания, появляющиеся во время сна.
Кожные симптомы
Коллагеновые структуры играют важную роль в поддержании функции защитных покровов. Эпидермис, состоящий из различных видов соединительной ткани, также может вовлекаться в патологический процесс. Кожа пациентов с синдромом Марфана не эластична, поэтому у них зачастую отмечается формирование растяжек без видимых на то причин. Данный дефект вызывает беспокойство у женщин, хотя и является исключительно косметологической проблемой.
Влияние на нервную систему
Головной и спинной мозг также окружены своеобразной капсулой из соединительной оболочки. У пациентов с синдромом Марфана отмечается постепенное ослабевание данной структуры. Подобная проблема сопровождается развитием неврологической симптоматики, характерной для дуральной эктазии. Отмечается появление слабости в тазовых конечностях, а также выраженной болезненности в брюшной полости.
Необходимые исследования
Постановка диагноза начинается с тщательного сбора анамнеза, особенно семейного. Наличие у прямых родственников пациента синдрома Марфана указывает на формирование данного недуга. Большое значение имеет и клиническая картина патологии, а именно наличие дефектов, возникающих вследствие нарушения строения соединительнотканных структур. Потребуется осмотр пациента кардиологом, окулистом, специалистом в области ортопедии. Для выявления мутации применяются методики генетического анализа, которые в диагностике синдрома Марфана занимают одно из лидирующих положений. В ряде случаев, особенно при подозрении на опасные осложнения, например, аневризму аорты, оправдано проведение УЗИ, а также КТ и МРТ. Визуальные методы применяются с целью создания фото внутренних органов для оценки их структуры и размеров.

Основные методы лечения
На сегодняшний день специфических способов борьбы с патологией не разработано. Терапия носит симптоматический характер, направлена на увеличение продолжительности жизни пациента за счет снижения риска опасных осложнений. Применяются как медикаментозные средства, так и более радикальные методы, вплоть до хирургического вмешательства. В ряде случаев лечебным эффектом обладают и народные рецепты. Важную роль играет также сбалансированное питание и соблюдение принципов здорового образа жизни. Тактику борьбы с недугом определяет врач на основании проведенных диагностических тестов.
Консервативные
Среди фармакологических средств применяются препараты следующих групп:
- Бета-адреноблокаторы, к которым относится «Метопролол», назначаются при наличии расширения аорты. Эти соединения способствуют снижению давления, за счет чего удается снять нагрузку с сосуда и предотвратить формирование разрыва, который способен привести к смерти пациента.
- Комплексные витаминно-минеральные добавки применяются с целью коррекции метаболических нарушений в костной ткани. Используются БАД, содержащие в составе магний, цинк, железо и другие микроэлементы.
- Антикоагулянты, например, «Гепарин», используются для профилактики осложнений, которые могут сформироваться в ходе прогрессирования заболевания. Подобная терапия широко применяется в тех случаях, когда в патологический процесс вовлекается сердечно-сосудистая система.
- При выявлении инфекционных поражений оправдано использование антибиотиков различных групп.
Народные рецепты
Лечить заболевание можно и нетрадиционными методами, но только после консультации с врачом. Использование подобных средств определяется симптомами, возникающими у пациента. Например, при выявлении признаков сердечной недостаточности готовят настой из березовых листьев. Если человек страдает от боли в суставах, широко применяются примочки на основе меда, спирта и скипидара.

Хирургическое вмешательство
При болезни Марфана проводятся различного рода операции. Они практикуются с целью устранения врожденных дефектов, а в ряде случаев и для спасения жизни пациента. При наличии выраженных пороков сердца, связанных с недостаточностью функции клапанов, осуществляется их протезирование. Хирургическое вмешательство зачастую необходимо людям с синдромом Марфана, у которых диагностировано существенное расширение аорты, поскольку данный дефект способен привести к гибели.
Оперативные техники используются также для коррекции зрения пациентов. В ряде случаев применяется оборудование, позволяющее осуществлять лечение при помощи лазера. Таким образом успешно исправляется миопия. Оправдан этот подход также при борьбе с глаукомой и катарактой. При помощи современных методов проводится и имплантация хрусталика, то есть замена старой структуры на новую.
Краткие сведения об известных людях, страдающих синдромом Марфана
Великий скрипач Николо Паганини страдал от данного генетического дефекта. С этим диагнозом жил и Авраам Линкольн, ставший президентом Соединенных Штатов. Известный писатель Ганс Христиан Андерсен также имел наследственное заболевание. Все эти люди отличались характерным высоким ростом и непропорциональностью телосложения.
Прогноз и профилактика
Течение патологии во многом определяется своевременностью проводимого лечения. На жизнь пациентов накладываются ограничения, например, мужчин с синдромом Марфана не призывают на службу в армию, поскольку всем больным не рекомендованы физические нагрузки. При этом само по себе поражение не может приводить к гибели. Летальный исход провоцируют формирующиеся на фоне генетической аномалии осложнения, поражающие сердце и другие органы.
Специфических средств профилактики синдрома Марфана не разработано. Поэтому для предотвращения появления на свет ребенка с данным дефектом родителям рекомендуется правильное планирование беременности. При наличии в семейном анамнезе случаев диагностики патологии врачи советуют проведение хромосомного анализа.
Изучение заболевания существенно продвинулись за последнее десятилетие. Было проведено рассмотрение патогенеза проблемы. Результаты указывают на участие в формировании симптомов именно множества вариаций мутации участка ДНК, ответственного за синтез фибриллина. Гаплоинтенсивность играет значимую роль в клинической картине синдрома Марфана, причем намного более заметную, чем просто угнетение производства белка. Отсутствие микрофибрилл в клетках соединительной ткани, которое также считается характерным признаком заболевания, формируется уже после появления ребенка на свет. Подобная постнатальная проблема обозначается термином «приобретенный эластолиз», то есть не относится к числу врожденных аномалий.
Понимание патогенеза позволило медикам продвинуться в контроле синдрома Марфана. Большинство предпринимаемых действий направлено, в первую очередь, на защиту аорты, поскольку именно данный сосуд больше всего страдает от нарушения строения соединительной ткани. Также его поражение связано с высоким риском развития опасных осложнений. В связи с этим современная концепция лечения синдрома связана с использованием бета-адреноблокаторов, регулярной последовательной визуализацией кардиальных структур. Широко применяются хирургические методики восстановления и замены участков аорты, позволяющие значительно продлить жизнь пациентов.
Отзывы
Анна, 23 года, г. Москва
У меня от рождения деформирована грудная клетка. Всегда была очень худой и высокой. Периодически болели суставы, особенно колени и пальцы. Еще в детстве врачи заподозрили синдром Марфана, но сделать генетический тест я решилась только сейчас. Предположительный диагноз подтвердился. Однако болезнь никак не беспокоит. Доктора рекомендовали регулярно проходить диспансеризацию.
Юрий, 47 лет, г. Серпухов
Сыну еще в школе поставили диагноз «синдром Марфана». Обратились к врачу с жалобами на плохую переносимость физических нагрузок. Доктора провели комплексное обследование. По типу телосложения заподозрили эту болезнь, которая позже подтвердилась в результате генетического теста. Теперь сын регулярно проходит диспансеризацию, сдает анализы, делает УЗИ сердца.
 Загрузка…
Загрузка…фото, симптомы, диагностика, лечение, наследование болезни
Синдром Марфана — это генетическое заболевание, при котором патологические изменения затрагивают соединительную ткань. Из-за этого у пациента развиваются нарушения в строении скелета и работе сердца, а также ухудшается зрение. Такая болезнь встречается одинаково часто и у мужчин, и у женщин. Патология отмечается в 1 случае на 10000 новорожденных. В дальнейшем болезненные изменения прогрессируют и без лечения существенно сокращают продолжительность жизни пациента.
Причины заболевания
Основной причиной синдрома Марфана являются врожденные изменения в гене FBN1. Он контролирует выработку в организме белка фибриллина. Это вещество делает соединительную ткань эластичной и прочной.
В организме больных этот белок продуцируется в очень малом количестве или совсем не вырабатывается. Его дефицит негативно сказывается на состоянии соединительной ткани. Она слишком сильно растягивается, становится слабой и плохо справляется даже с небольшими физическими нагрузками. Из-за этого у людей с синдромом Марфана неправильно развивается скелет, отмечаются серьезные нарушения в работе сердечных клапанов. Вследствие подвижности хрусталика глаза наблюдается ухудшение зрения. Дефекты в соединительной ткани происходят еще во время внутриутробного периода. В течение жизни они прогрессируют. Без терапии это заболевание может привести к ранней смерти пациента.
Как наследуется патология?
Наследование синдрома Марфана происходит по аутосомно-доминантному типу. Если болен один родитель, то он может передать патологию ребенку в 50% случаев. Если же этим синдромом страдают и отец, и мать, то вероятность рождения больного малыша увеличивается до 75-100%.
Чаще всего патология передается от больных родителей. Но бывает и так, что отец и мать здоровы, однако ребенок страдает синдромом Марфана. Это отмечается в 15% случаев. Причины такой случайной мутации гена неизвестны. Однако специалистами установлено, что риск рождения ребенка с такой патологией увеличивается, если возраст отца превышает 35-40 лет. Возраст матери в этом случае не влияет на мутацию гена.
Признаки поражения опорно-двигательного аппарата
Изменения в скелете являются одним из ведущих симптомов синдрома Марфана. Опытный врач может предположить наличие этой патологии уже во время осмотра по внешнему виду больного.
Люди с этим заболеванием обычно имеют высокий рост. У них тонкие кости с повышенной гибкостью в суставах и сухожилиях. При большом росте пациенты обычно имеют низкий вес и выглядят высокими и худощавыми. Кости удлиняются из-за чрезмерного растяжения соединительной ткани при синдроме Марфана. Фото больного можно увидеть ниже.

Конечности больных непропорционально длинные и тонкие. Некоторые пациенты, имея высокий рост и большой размах рук, пытаются заниматься спортом. Однако при такой патологии физические нагрузки категорически противопоказаны. Соединительная ткань из-за недостатка фибриллина становится очень слабой и может не выдержать напряжения. При высоком росте больные не отличаются большой физической силой, напротив, их кости и мышцы плохо справляются с нагрузкой.
Другим признаком патологии являются непропорционально длинные пальцы. Они обладают повышенной гибкостью. Такой симптом называют «паучьими пальцами», или арахнодактилией, это одно из характерных внешних проявлений синдрома Марфана. Фото кисти больного можно увидеть ниже.
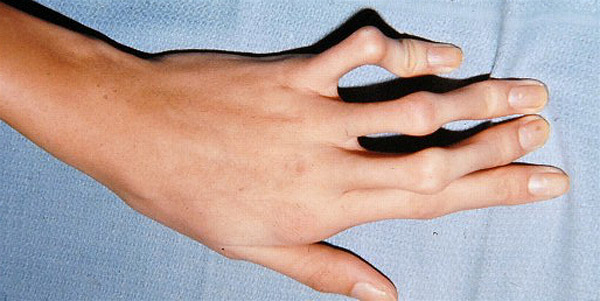
У страдающих этим синдромом лицо имеет длинную и узкую форму. Есть изменения в небе, оно высокое и изогнутое. Из-за этого у больного ребенка могут неправильно расти зубы. У взрослых меняется голос, появляется хрипота.
Жировая ткань у пациентов мало развита, из-за этого они имеют пониженную массу тела и выглядят очень худыми при высоком росте. Мышцы слабые, плохо выдерживают нагрузку.
У больных отмечается плоскостопие, искривления позвоночника и деформации костей туловища. Вдавленная или выступающая грудная клетка — частое проявление синдрома Марфана. На фото можно увидеть деформацию костей у больного.
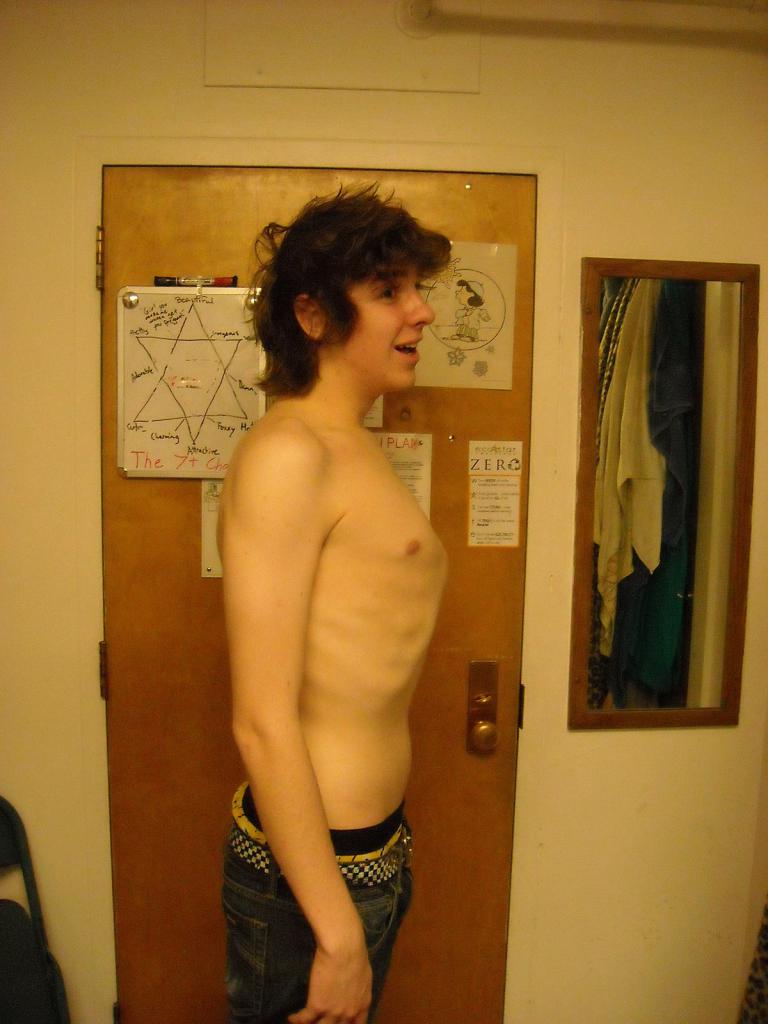
Ухудшение зрения
Из-за большой растяжимости соединительной ткани повышается подвижность хрусталика глаза. Нередко происходит вывих или подвывих этого органа. При осмотре у окулиста определяется сильное смещение хрусталика. Это негативно сказывается на зрении. У пациентов с синдромом Марфана выявляются следующие офтальмологические нарушения:
- близорукость;
- глаукома;
- катаракта.
Эти заболевания возникают еще в детстве. Часто ребенку с малых лет приходится носить очки. Опасным осложнением смещения хрусталика может стать отслойка сетчатки, которая приводит к сильному ухудшению зрения.

Симптомы со стороны сердца
Поражение сердца является наиболее опасным признаком синдрома Марфана. Кардиологические патологии нередко становятся причиной летального исхода из-за разрыва аорты. У пациента отмечаются следующие болезненные изменения:
- расширение и аневризма аорты;
- нарушение работы сердечных клапанов;
- увеличение сердца;
- нарушение проводимости миокарда.
Из-за этих отклонений наблюдаются боли в груди по типу стенокардии, одышка, аритмия, тахикардия, ощущение постоянной усталости. Иногда дети, унаследовавшие этот синдром, рождаются с пороками сердца, что приводит к гибели на первом году жизни.
Поражение нервной системы
При синдроме Марфана симптомы со стороны центральной нервной системы связаны с набуханием мешочка (оболочки), который окружает спинной мозг. Это образование также состоит из соединительной ткани, которая патологически изменена. Врачи-невропатологи называют такое нарушение дуральной эктазией. Сначала человек испытывает лишь небольшой дискомфорт. Но со временем патология прогрессирует, и усиливается давление оболочки на нижние отделы спинного мозга. Больного беспокоят частые боли в животе, а также слабость и онемение ног.
Изменения в легких
Патологические изменения в легких отмечаются не всегда. Однако эластичность соединительной ткани нарушена и в органах дыхания. В результате этого повышается риск возникновения пневмоторакса, дыхательной недостаточности и эмфиземы легких. У больных нередко происходит спонтанная остановка дыхания (апноэ) в ночное время.
Другие проявления
У пациента появляются растяжки на коже, которые никак не связаны с изменением массы тела. Это является лишь косметическим недостатком и обычно не доставляет никакого беспокойства.
Однако пациенты очень склонны к образованию грыж в паховой и брюшной области, которые часто рецидивируют, несмотря на лечение.
Этот синдром нередко сопровождается эндокринными нарушениями, у больных повышен риск возникновения сахарного диабета и акромегалии. Из-за растяжения соединительной ткани происходит опущение мочеполовых органов. Пациенты часто получают травмы: вывихи и растяжения сухожилий.
Общее самочувствие
При этой генетической патологии больные чувствуют себя постоянно уставшими. Они утомляются даже от незначительного физического напряжения. Мускулатура их слабо развита, после нагрузки пациенты ощущают боли в мышцах. Такие признаки связаны с поражением соединительной ткани.
При психоэмоциональном напряжении могут возникать головные боли по типу мигрени, астения и понижение АД.
Страдает ли умственное развитие
У больных детей не отмечается задержки умственного развития. Пациенты с синдромом Марфана имеют нормальный интеллект. В некоторых случаях их уровень IQ даже выше среднего. Однако у больных иногда наблюдаются признаки повышенной нервозности: излишняя эмоциональность, плаксивость, раздражительность.

Диагностика
Врач может предположить заболевание по внешнему виду пациента. Однако высокий рост, худощавая комплекция и плохое зрение не всегда говорят о данной патологии. Эти признаки наблюдаются и при других генетических нарушениях, и у здоровых людей. Специалист обязательно расспрашивает больного о случаях этой наследственной аномалии в семье, в частности, у родителей.
Для диагностики синдрома Марфана необходимо пройти обследование у ортопеда, кардиолога, офтальмолога и генетика, а также сделать ЭКГ. Это поможет оценить состояние костей, глазного аппарата и сердца. Диагноз ставят, если у человека отмечается хотя бы один из следующих симптомов:
- деформация грудных костей;
- смещение хрусталика;
- искривление позвоночника;
- патологические изменения в аорте;
- удлинение и патологическая гибкость пальцев.
А также у человека при этом должны наблюдаться хотя бы два из следующих признаков:
- нарушение работы сердечных клапанов;
- пневмоторакс;
- плоскостопие;
- высокий рост;
- чрезмерная подвижность сухожилий и суставов;
- близорукость;
- растяжки на теле.
Таким образом, диагноз ставят по совокупности проявлений заболевания. Кроме этого, существует специальный анализ на синдром Марфана. У пациента берут кровь и проводят поиск мутаций в гене FBN1. Однако это довольно дорогостоящее исследование, его проводят в центрах молекулярной генетики.

Консервативное лечение
В современной медицине нет таких методов, которые могли бы исправить «поломку» гена. Поэтому лечение синдрома Марфана может быть только симптоматическим. Назначают следующие медикаменты, которые помогают скорректировать патологические изменения в организме:
- Бета-адреноблокаторы («Атенолол», «Обзидан» и другие). Их применяют при расширении аорты до 4 см для предотвращения кардиологических осложнений. Также используют и другие сердечные препараты (ингибиторы АПФ, антагонисты кальция).
- Препарат «Лозартан». Это лекарство стало использоваться в терапии болезни Марфана относительно недавно. Оно относится к блокаторам рецепторов ангиотензина. Препарат замедляет расширение аорты. По исследованиям медиков это лекарство действует более эффективно, чем бета-адреноблокаторы.
- Антибиотики и коагулянты. Их назначают обычно после кардиологических операций, для профилактики тромбов и эндокардита.
- Препараты для нормализации коллагена. С этой целью назначают антиоксиданты, витаминные комплексы и биодобавки: «Коллаген ультра», «Элькар», «Лимонтар», «Рибоксин», средства с коэнзимом, витамины Е и группы В.
- Ноотропы. Их назначают при слабости и астении. Чаще всего применяют «Пирацетам».
Больной должен находиться под наблюдением нескольких врачей (генетика, кардиолога, ортопеда, офтальмолога), регулярно проходить осмотры и необходимые обследования. Коррекция зрения при миопии проводится с помощью подбора очков или контактных линз.
Пациентам с болезнью Марфана показаны физиотерапевтические процедуры. Полезно проведение магнитотерапии на суставы, электросна, упражнений ЛФК. Раз в год рекомендуется лечение в санатории для больных с патологиями опорно-двигательного аппарата.
Хирургические операции
При этом синдроме часто приходится прибегать к кардиохирургическим вмешательствам. Это позволяет существенно увеличить выживаемость больных. До применения операций на сердце продолжительность жизни пациентов с этим синдромом была небольшой. Люди доживали только до среднего возраста (40-50 лет). В настоящее время кардиохирургическое лечение увеличило жизнь пациентов примерно на 20 лет.
Нередко приходится делать операции на аорте. Они показаны при расширении этого сосуда более 5 см. Также осуществляют протезирование митрального клапана.
Операции на сердце делают чаще всего при таком синдроме, так как этот орган сильно страдает от дефектов соединительной ткани. Однако проводят и другие хирургические вмешательства, такие как:
- Торакопластика. Эту операцию делают при деформации грудной клетки. Она позволяет избавиться от дефекта строения скелета. Проводится частичная резекция нескольких ребер, после этого грудина выглядит более ровной.
- Экстракция хрусталика. Такую операцию на глазах проводят при ранней катаракте и глаукоме.
- Стабилизирование позвоночника с помощью металлических пластин. Операцию проводят в тяжелых случаях сколиоза.
- Эндопротезирование. У больных часто отмечаются патологии костей. По этой причине иногда приходится заменять пораженный сустав на протез.
- Удаление аденоидов и гланд. Эти вмешательства необходимо провести в детстве. Ребенок, страдающий данным синдромом, очень подвержен простудным заболеваниям.
Кроме этого, беременным женщинам с болезнью Марфана показана операция кесарева сечения. Из-за слабости мышц и проблем с сердцем им очень трудно рожать самостоятельно.
Прогноз
При этом наследственном недуге необходимо комплексное лечение всех органов, которые затронуты болезнью. Без терапии продолжительность жизни больных составляет не более 40-45 лет. Смерть наступает от тяжелых нарушений работы сердца и сосудов.
Хирургическая коррекция патологических изменений позволяет значительно увеличить жизнь больных. При регулярном лечении и врачебном наблюдении пациенты могут дожить до пожилых лет. Кроме этого, после кардиохирургических операций возрастает трудоспособность больного.
Молодые женщины, страдающие этим синдромом, часто интересуются: можно ли им беременеть. Шанс родить здорового малыша существует при условии, что отец ребенка здоров. Однако необходимо помнить, что риск передачи заболевания в этом случае составляет 50%. К тому же вынашивание плода создает огромную нагрузку на сердечно-сосудистую систему, которая и так страдает при этом недуге. Поэтому при подобной генетической патологии беременеть не рекомендуется.
Профилактика
На сегодняшний день не разработана специфическая профилактика синдрома Марфана. Заболевание является генетическим и предотвратить его нельзя. Можно лишь пройти пренатальную диагностику во время беременности. Если один из будущих родителей страдает этой болезнью, то необходима консультация генетика.
Если у человека уже отмечаются признаки патологии, то необходимо избегать физических нагрузок. Больным не следует заниматься спортом и выполнять тяжелую работу. Такие пациенты должны регулярно проходить медицинское обследование. Дети с этим недугом освобождаются от занятий физкультурой, однако им показано выполнение упражнений ЛФК.
Болезнь арахнодактилия (паучьи пальцы): это паукообразные пальцы | Ревматолог
В числе серьезных генетических заболеваний находится синдром Марфана, которым страдает один человек из 10-20 тысяч. Им болели такие всемирно известные личности, как Авраам Линкольн, Никколо Паганини, Корней Чуковский и Ганс Христиан Андерсен.
У больного этим синдромом присутствуют различные симптомы: арахнодактилия (удлиненные кисти, паукообразные пальцы рук и ног), множественные костно-суставные аномалии, деформация грудной клетки, нарушения сердечно-сосудистой системы и т.д. В этой статье речь пойдет об одном из них — арахнодактилии, ее проявлениях, методах диагностики и способах лечения.
Что такое арахнодактилия
Это вид наследственной патологии человека, который проявляется аномально длинными и тонкими пальцами рук, их чрезмерной подвижностью, вплоть до сгибания на 180 градусов к тыльной стороне ладони. В народе это заболевание приобрело название болезни «,паучьи пальцы»,.
После описанного впервые синдрома Марфана в 1876 году было выявлено, что «,паучьи пальцы», могут быть признаком не только этой патологии, но и некоторых других генетических заболеваний и мутаций, нарушения синтеза белка.
Причины появления
Достоверные и точные причины появления этой патологии у человека до сих пор неизвестны. Большая часть исследователей придерживается той версии, что заболевание связано с нарушением строения коллагеновых волокон и на 75% передается наследственным путем.
Остальные 25% связывают с различными генетическими мутациями, которые могут произойти спонтанно в период зачатия как в яйцеклетке, так и в сперматозоиде.
Формы проявления заболевания, общие симптомы и признаки
Выделяют три основных вида патологических нарушений, для которых характерен симптом «,паучьих пальцев»,:
- Синдром Марфана. Основной вид патологии, передающийся по наследству по аутосомно-доминантному признаку. Сопровождается ярко выраженной деформацией костно-мышечной системы и осложнениями на сердечно-сосудистой системе.
- Гомоцистинурия. Генетическое заболевание, которое передается по аутосомно-рецессивному типу. Редкая патология, встречается примерно у одного человека на 200-300 тысяч.
- Непредвиденные генные мутации, вызывающие патологии различного характера.
Внешние симптомы больного арахнодактилией:
- длинные и тонкие конечности и пальцы,
- выраженный сколиоз, кифосколиоз,
- воронкообразная, суженная, удлиненная грудная клетка,
- талия, расположенная выше нормы,
- аномально высоко сформированная надколенная чашечка,
- деформированные кости таза,
- узкий удлиненный череп,
- неразвитая мышечная система, выраженная худоба,
- широко расставленные глаза,
- высокий свод неба,
- неправильное развитие глаз и другие симптомы.
Основные признаки болезни:
- многочисленные болезни клапанов сердца и аорты,
- гипоплазия легких,
- офтальмологические заболевания (близорукость, косоглазие, катаракта, подвывих хрусталика и др.),
- поражение артерий,
- остеопороз,
- разболтанность суставов,
- припадки,
- могут присутствовать отклонения в психике.
Интересно! Характерный для арахнодактилии выброс адреналина способствует развитию выдающихся умственных способностей и одаренности. Некоторые больные могут отличаться высокой силой духа и сильными волевыми качествами.

Методы диагностики
Внешние проявления заболевания легко диагностируются при помощи визуального осмотра пациента, замера длины конечностей и гибкости суставов. При обнаружении первых признаков назначается комплексное обследование для выявления сопутствующих заболеваний, патологических изменений, нарушений работы внутренних органов.
Для этого применяют следующие исследования:
- рентген позвоночника, груди и таза,
- МРТ, КТ костей и суставов,
- сцинтиграфия.
Для выявления нарушений работы сердечно-сосудистой системы назначают:
- УЗИ сердца и сосудов,
- томографию артерий и аорты,
- анализы крови.
При подозрении на гомоцистинурию проводят:
- МРТ головного мозга,
- КТ черепа и позвоночника,
- электроэнцефалографию,
- реоэнцефалографию.
Также необходимо обследовать органы зрения у офтальмолога, для чего проводят различные оптические и УЗИ-исследования.
Лечение
Лечение для каждого больного арахнодактилией подбирается строго индивидуально. Это связано с тем, что наличие симптомов или патологий проявляется по-разному. План лечебных и профилактических мероприятий подбирается в зависимости от особенностей протекания болезни. Как правило, основой становятся ортопедические мероприятия, направленные на коррекцию всех имеющихся деформаций.
Полностью устранить болезнь на сегодняшний день нельзя, но есть возможность значительно облегчить состояние больного.
В настоящее время разработан метод коллагенонормализующей терапии, благодаря которому корректируется метаболизм компонентов соединительной ткани, усиливается регенерация хрящей при хирургическом вмешательстве.
Если у человека проявляются заболевания сердечно-сосудистой системы или глаз, то в соответствии с показаниями назначается необходимая терапия по устранению или облегчению симптомов.
При наличии патологий грудной клетки проводится торакопластика – операция по достижению нормального положения грудной клетки и уменьшению ее размеров.
Медикаменты
В зависимости от симптоматики могут быть назначены разнообразные медикаменты, но основными лекарствами являются антиоксиданты и энерготропные препараты: «,Рибоксин»,, «,Коэнзим»,, «,Пирацетам»,, «,Лимонтар»,, витамины группы В и С.
ЛФК
Вне зависимости от причины возникновения заболевания пациенты находятся на постоянном диспансерном наблюдении, посещают ЛФК и массаж.
Важно! ЛФК при арахнодактилии можно применять только при невыраженных нарушениях опорно-двигательного аппарата и исключительно под наблюдением специалиста.
Массаж

Массажные процедуры при арахнодактилии назначаются индивидуально в зависимости от особенностей и патологий пациента, проводятся в комплексе и направлены на лечение и облегчение симптомов патологии.
Массаж выполняется только специально обученными медицинскими работниками, имеющими соответствующую квалификацию.
Заключение
Арахнодактилия — тяжелое и непредсказуемое заболевание, причины появления которого до сих пор не изучены до конца. Чтобы жить с такой патологией, понадобится пройти немало испытаний, проявить силу духа и терпение.
Помимо описанных в статье методов лечения, больному необходимо в качестве профилактики и повышения жизненного тонуса нормализовать сон и обязательно соблюдать все рекомендации врача по питанию и физической активности.
Арахнодактилия (синдром паучьих пальцев) — симптомы, лечение и фото

Арахнодактилия – клинический синдром, возникающий при некоторых наследственных заболеваниях. Проявляется деформацией пальцев кисти: пальцы длинные, тонкие, характерно изогнутые. Как правило, арахнодактилия сочетается с удлинением всех трубчатых костей, другими деформациями скелета, патологическими изменениями со стороны глаз и сердечно-сосудистой системы. Реже арахнодактилия возникает изолированно, без поражения других органов и систем. Диагноз выставляется на основании комплексного обследования, включающего в себя изучение генеалогических данных, оценку нервно-психического и физического состояния больных с использованием специальных шкал и индексов, а также ряд анализов и инструментальных тестов для выявления патологии со стороны внутренних органов. Лечение симптоматическое.
Арахнодактилия
Арахнодактилия (в переводе с латыни «паучьи пальцы») – синдром, который наблюдается при некоторых наследственных болезнях, связанных с нарушением обмена веществ. Первое описание данной патологии, сочетающейся с другими характерными изменениями скелета, относится к 1876 году. Через 20 лет французский педиатр Марфан опубликовал исследования, в которых описывал аналогичные нарушения у пятилетней девочки. Заболевание стало известно под названием «синдром Марфана». В последующем было установлено, что арахнодактилия является наследственной патологией соединительной ткани и может обнаруживаться не только при синдроме Марфана, но и при гомоцистинурии, а также при некоторых других редких наследственных синдромах.
Причины арахнодактилии
Чаще всего арахнодактилия наблюдается при синдроме Марфана и гомоцистинурии. Существует также ряд так называемых марфаноподобных синдромов – наследственных болезней, которые сопровождаются арахнодактилией и внешне напоминают синдром Марфана, но отличаются по проявлениям со стороны внутренних органов. К числу болезней, сочетающихся с арахнодактилией, относится синдром дилатации и расслоения аорты, синдром эктопии хрусталиков и некоторые другие редкие генетические заболевания.
Предполагается, что причиной развития синдрома Марфана и других синдромов, при которых возникает арахнодактилия, является нарушение синтеза белка, обусловленное схожими генетическими мутациями. При синдроме Марфана отмечается аутосомно-доминантный тип наследования, при гомоцистинурии – аутосомно-рецессивный. Кроме того, арахнодактилия может являться результатом спонтанных генетических мутаций.
Симптомы арахнодактилии
Арахнодактилия проявляется характерными изменениями формы и размеров кисти. Пальцы непропорционально тонкие по сравнению с ладонью, чрезмерно длинные, изогнутые, с типичной деформацией в области межфаланговых суставов. Все перечисленные проявления придают кистям больного своеобразный вид, из-за которого патология получила наименование «паучьих пальцев». У некоторых больных наряду с необычным внешним видом обнаруживается чрезмерная подвижность пальцев – один или несколько из них могут отклоняться на 180° в тыльную сторону. Обычно удлинение пальцев сопровождается другими патологическими изменениями со стороны скелета, глаз и внутренних органов. Характер изменений определяется типом наследственного заболевания.
При синдроме Марфана арахнодактилия сочетается с другими изменениями скелета: все трубчатые кости удлиненные, руки и ноги непропорционально тонкие и длинные. Из-за слабости связочного аппарата часто возникает избыточная подвижность суставов. Мышцы атрофированы и почти незаметны под кожей, жировой слой практически не выражен. Голова увеличена в размере, череп удлинен, затылок уплощен, лоб высокий, с ярко выраженными лобными буграми. Нижняя челюсть выпячена кпереди или, наоборот, слабо выражена и выглядит недоразвитой. Позвоночник искривлен, таз плоский, грудная клетка воронкообразная или килеобразная. Наблюдается выраженное плоскостопие.
Наряду с арахнодактилией и другими изменениями костно-мышечной системы выявляется патология со стороны глаз и сердечно-сосудистой системы. Склеры голубоватые, отмечается близорукость, характерен подвывих хрусталиков кверху. Часто обнаруживается аневризма аорты и различные пороки сердца. Степень выраженности различных патологических изменений при арахнодактилии может значительно варьироваться – от слабой, с отсутствием ряда или большинства признаков, до тяжелой, с нарушением способности к самообслуживанию и неблагоприятным прогнозом для жизни.
Диагностика арахнодактилии
Диагноз того или иного заболевания, сопровождающегося арахнодактилией, выставляется на основании внешнего осмотра и ряда инструментальных исследований. При наличии показаний назначается рентгенография таза, рентгенография позвоночника, рентгенография грудной клетки, сцинтиграфия, КТ костей и суставов, МРТ костей и суставов. При необходимости больного арахнодактилией направляют на консультации к неврологу, кардиологу и офтальмологу. Кардиолог может назначить УЗИ сердца и УЗИ артериальных сосудов, томографию сосудов и ряд других исследований. Невропатолог проводит специальные тесты и может направить пациента на МРТ головного мозга, КТ позвоночника и КТ черепа, электроэнцефалографию, реоэнцефалографию и пр. Офтальмолог выполняет осмотр структур глаза, при наличии показаний измеряет внутриглазное давление, назначает оптические, ультразвуковые и другие исследования.
В процессе обследования осуществляют дифференциальную диагностику арахнодактилии при гомоцистинурии и синдроме Марфана. Для гомоцистинурии характерны судороги и слабоумие, при синдроме Марфана интеллект сохранен, судорожный синдром отсутствует. При гомоцистинурии отмечается подвывих хрусталиков книзу, поражение глаз носит прогрессирующий характер, со временем развивается вторичная глаукома. У больных синдромом Марфана обнаруживается подвывих хрусталиков кверху, близорукость протекает более благоприятно, вторичная глаукома не развивается.
При гомоцистинурии наблюдается остеопороз, менее выраженное плоскостопие и умеренная или незначительная избыточная подвижность суставов, выявляется поражение артерий среднего калибра (почечных, мозговых, венечных), при синдроме Марфана остеопороз отсутствует, плоскостопие и избыточная подвижность суставов выражены более ярко, превалирует патология сердца и крупных сосудов. Гомоцистинурия чаще диагностируется у блондинов со светлой кожей и голубыми глазами, синдром Марфана – у достаточно смуглых брюнетов. При гомоцистинурии в моче и биохимическом анализе крови определяется повышенное содержание метионина и снижение уровня цистина, обнаруживается гомоцистеин, при синдроме Марфана перечисленные изменения отсутствуют.
Лечение арахнодактилии
Лечение арахнодактилии симптоматическое. Больным с арахнодактилией при синдроме Марфана для нормализации обмена веществ назначают большие дозы витамина С и анаболические стероиды. Пациентам с арахнодактилией при гомоцистинурии прописывают специальную диету с ограничением количества животных белков, большие дозы витаминов группы В и малые дозы препаратов, препятствующих свертыванию крови (аспирин). Больные с арахнодактилией вне зависимости от причины ее возникновения находятся на постоянном диспансерном наблюдении, по показаниям направляются на ЛФК, массаж и санаторно-курортное лечение. При патологии, угрожающей жизни пациента и существенно ухудшающей его состояние, проводятся различные операции: протезирование сердечных клапанов, протезирование аорты, хирургическая коррекция воронкообразной и килеобразной грудной клетки и т. д.
Арахнодактилия (паучьи пальцы): причины заболевания, способы лечения
Арахнодактилия – это заболевание, известное в народе как «паучьи пальцы», для него характерны анатомические изменения и деформирование конечностей: удлинение и истончение фаланг пальцев, изменение конфигурации хрящей, суставов и т.д. При арахнодактилии пальцы больного внешне напоминают лапки паука, они имеют неправильную форму и пропорции.
Хотя в некоторых случаях длинные и тонкие пальцы могут быть попросту анатомической особенностью человека, являясь вполне нормальным явлением, не связанным с медицинской патологией.
Причины возникновения арахнодактилии
Причины развития арахнодактилия, по сей день, не установлены. Поэтому, все заявления врачей и предположения по данному поводу нельзя считать чем-либо большим, чем просто гипотезами.
Наиболее вероятна версия о нарушении строения коллагеновых волокон, влекущем за собой ряд неблагоприятных процессов. Данный дефект нельзя считать распространенным. Арахнодактилия встречается лишь у 1 из 5000 человек. При этом, более чем в 75% случаев, заболевание носит наследственный характер, то есть, передается детям от ближайших родственников.
Что касается оставшихся 25%, то болезнь, в данном случае, возникает без каких-либо видимых причин, у ребенка, чьи родители здоровы. Возможно, виной тому спонтанные генетические мутации, происходящие в яйцеклетке матери, или сперматозоиде будущего отца ребенка. Процесс мутации происходит в 15-й хромосоме и затрагивает ген белка.

Среди возможных причин развития арахнодактилии:
- болезни обмена белков;
- синдром Марфана, являющейся аутосомно-доминантным заболеванием, относящимся к группе наследственных патологий соединительных тканей;
- иные генетические изменения.
Таким образом, если кто-либо из родителей болен арахнодактилией, то сообщить об этом врачу следует еще на этапе плакирования беременности. Тщательное обследование поможет выявить наследование ребенком аномалии и в период внутриутробного развития.
Клиническая картина заболевания
Клинически арахнодактилия представляется в виде аномалии строения и удлинении косных тканей. Визуально, это наиболее заметно по форме кистей рук и стоп.
Хотя некоторые изменения заметны уже в первые дни жизни ребенка, в полную меру болезнь проявляет себя лишь к 3-4 годам жизни. Длина тела новорожденных изрядно превышает среднестатистические нормы. Удлиненные и истонченные пальцы на руках и ногах часто имеют причудливую форму (искривлены), а сухожилья, напротив, укорочены и развиты довольно слабо.
Конечности тонкие и длинные, предплечье и голени увеличены, аномально высоко сформирована надколенная чашечка. Все это в целом влечет за собой значительные изменения пропорций тела.
Таким образом, туловище больного сильно искажено:
- талия находится на несколько сантиметров выше нормы;
- грудная клетка узкая, воронкообразно вдавлена;
- наличие сколиоза;
- узкая и удлиненная форма черепа;
- поражен мышечно-связочный аппарат;
- кости таза деформированы.

Часто у больных арахнодактилией наблюдаются врожденные вывихи бедра, привычные вывихи плечевых костей, надколенной чашечки, а также радио-ульнарных суставов. Деформирована пяточная кость. Все суставы и хрящи чрезвычайно подвижны, при этом, суставные сумки и сухожилия чрезмерно растянуты.
Иссушены мышцы, их тонус ниже положенных норм. Слабо развита подкожно-жировая клетчатка. Как правило, больные с данным диагнозом имеют еще и ряд сопутствующих отклонений в развитии, либо заболеваний внутренних органов.
Более трети больных имеют те или иные проблемы со стороны сердечно-сосудистой системы: врожденные пороки сердца (диффект в строении перегородок), аневризм аорты, коарктацию аорты, незаращения боталлова протока.
Данному заболеванию часто сопутствует врожденная гипоплазия легкого. Более чем у 50% больных наблюдается ряд проблем со стороны офтальмологии: косоглазие, нистагм, высокий уровень близорукости, катаракта, гидрофтальмия, колобома и т.д.
Степень поражения
Тяжесть заболевания, как и продолжительность жизни больного полностью зависят от степени и масштабов поражения. Изменения в строении скелета не значительно влияют на дальнейшую жизнедеятельность. Больной арахнодактилией может дожить вплоть до глубокой старости, при отсутствии, или малом количестве сопутствующих отклонений и поражения жизненно-важных органов и тканей.
В случае сочетания костных поражений с пороками в развитии сердечно-сосудистой системы, а также тканей легкого, дальнейший прогноз может быть определен лишь в соответствии со степенью выраженности самих пороков.
Лечение арахнодактилии
Лечение арахнодактилии и дальнейший план действий подбирается на основе собранного анамнеза.
Больной проходит тщательный медицинский осмотр. Ему задается ряд вопросов об истории болезни:
- в каком возрасте пациент обнаружил изменения строения пальцев рук;
- что включает в себя семейная история болезни;
- наличие случаев ранней смерти родственников, больных арахнодактилией;
- основная причина смерти;
- иные наследственные заболевания;
- наличие тех или иных симптомов у пациента и т.д.
Данное диагностическое тестирование, как правило, не проводится, если заболевание не носит наследственный характер.
Лечение арахнодактилии, как правило, симптоматическое. Оно направлено на ортопедическую коррекцию деформаций, которые вторично развились на фоне основного страдания.

Вправление вывихов, вытяжение, редрессация костей рук и т.д. По мере взросления больного и дальнейшего роста конечностей, производят коррекцию развития мышечно-связочного аппарата, а также костно-суставного аппарата. В отдельных случаях приходится прибегать к оперативному вмешательству, например, в случае необходимости удаления сухожилий. Обычно операция показана лишь в том случае, если скорректировать ситуацию каким-либо иным способом не представляется возможным.
При выраженных патологиях развития тазобедренных либо коленных суставов, приводящих к ряду функциональных нарушений (хромоте, косолапости и т.д.), а также при возникновении интенсивных болевых ощущений, производится корригирующая остеотомия.
В случае развития плоскостопия, лечение предусматривает постоянное ношение ортопедической обуви и специальных вкладышей.
Назначить актуальное, в данном конкретном случае, лечение помогают данные, полученные путем проведения рентгенодиагностики. Таким образом, удается выявить патологии со стороны трубчатых костей, суставов, внутренних органов, строения позвоночника и т.д.
В целом, нет одного единого, подходящего для всех больных, метода лечения арахнодактилии. План необходимых мероприятий подбирается очень индивидуально, в соответствии с особенностями протекания болезни, степенью поражения внутренних органов, наличием тех или иных патологий.
Само же лечение подразумевает поддержание в жизнеспособном состоянии всех органов и систем. Полностью вылечиться от арахнодактилии, на сегодняшний день, не представляется возможным, поэтому действия врачей направлены более на облегчение состояние человека и обеспечение его нормальной жизнедеятельности.
Синдром «паучьи пальцы» или генетика — дело тонкое

Комментариев пока нет. Будь первым! 1,836 просмотров
У человека много генетических заболеваний, одним из таких проявляется синдром Марфана, который еще называется «паучьи пальцы». Основные симптомы синдрома Марфана связаны с нарушениями со стороны соединительной ткани. У больных людей нарушается производство белков в организме, из них потом синтезируется соединительная ткань. Болезнь прогрессирует, в результате чего у людей развиваются тяжелые нарушения, приводящие к инвалидности. Заболевание носит генетический характер, передается по наследству.
Немного истории
Первым описал болезнь, которая впоследствии получила имя Марфана врач Вильямсон в 1876 году. А название болезнь получила в честь педиатра из Франции по имени Марфан, который по прошествии двух десятилетий наблюдал ребенка, страдающего этой патологией. Известно, что болели известные люди, к примеру, Лесли Хорнби, которая стала прототипом эталона модельной индустрии. Также известные А. Линкольн, президент США, скрипач Паганини, как оказалось, имели синдром Марфана.
Потом по мере изучения стало известно: болезнь носит генетический характер, передается по наследству. Достаточно быть носителем синдрома патологического гена Марфана одному из родителей, чтобы потомство было больным. При отсутствии адекватного лечения синдрома Марфана, в том числе оперативного, прогноз неблагоприятный. Если не предпринимать мер, человек может не дожить до тридцати лет.
Симптоматика болезни
Симптомы сопровождаются поражением многих внутренних органов, поскольку соединительная ткань есть везде. Наиболее часто люди страдают нарушениями со стороны опорно-двигательного аппарата, сердечно-сосудистой, дыхательной, центральной нервной системы, нарушается зрение.
Симптомы проявляются астеническим типом телосложения: при высоком росте, свыше 180 сантиметров, очень маленькая масса тела, не больше 50 килограммов. Помимо этого, у людей очень длинные пальцы рук, ног, которые называют «паучьими пальцами». Деформируется ось позвоночника в виде кифосколиоза, грудная клетка становится килевидной, присоединяется плоскостопие. Лицо становится узким, небе высокое, готическое. Для людей с синдромом Марфана это наиболее распространенные симптомы, могут встречаться в отдельности или все вместе.
Синдром Марфана имеет типичное проявление: антимонголоидный разрез глаз, нос крупный, уши чрезмерно большие, расположены низко, лицо напоминает птицу. У новорожденного ребенка синдром Марфана проявляется только аномальным ростом, слишком длинными пальцами. Остальные симптомы проявляются на поздних сроках, как правило, на протяжении первых пяти или семи лет жизни.
Люди, страдающие заболеванием, имеют патологию со стороны сердца, сосудов. Развивается пролапс в области митрального клапана, аневризма (выпячивание) самого крупного сосуда – аорты. Вышеуказанные нарушения выявляются в течение первого или второго года жизни ребенка. Аорта увеличивается в диаметре, постепенно она достигает критического диаметра (более 6 см).
С возрастом, примерно от 16 до 45 лет, проявляется в полной мере. Самым грозным осложнением (оно способно возникнуть в любой момент) считается постоянное расслоение стенок аорты, при синдроме Марфана оно прогрессирует, постепенно распространяясь по всей длине сосуда, в отходящие ветви. Результатом является летальный исход: аорта может разорваться в любой момент, став причиной массивного кровотечения.
Часто люди, имеющие синдром Марфана, страдают патологией легких, бронхов. Причиной является деформированная грудная клетка из-за изменений соединительной ткани, которая входит в состав ткани дыхательной системы. Основные симптомы дополняет нарушение дыхания в качестве спонтанного пневмоторакса, не редкость такое состояние, как легочная эмфизема, инфаркт легкого встречается от 10% до 75 % при синдроме Марфана. Описаны ситуации врожденной аномалии легочной доли, булезной эмфиземы поликистоз, двухсторонних бронхоэктазов.
Со стороны зрения у людей, имеющих синдром Марфана, развивается смещение хрусталика глаза. Причиной подобного состояния является слабость связки, которая его удерживает. В дополнение у человека развивается близорукость или дальнозоркость, выраженная в сильной степени. Смещение хрусталика подтверждается в срок от первого до пятого года жизни, порой родители ничего не знают до 7 лет, все становится известно, когда ребенок идет в школу. Вторичным состоянием считается глаукома, катаракта, может отслоиться сетчатка. Изменения наблюдаются от 15 до 40 лет, ясно указывают на наличие патологии Марфана.
Человек, больной синдромом Марфана, имеет сниженное умственное развитие. Ребенок плаксив, раздражителен, имеет завышенную самооценку. Снижена устойчивость к физической нагрузке, в результате ее наступает быстрое переутомление мышц, боль. Во время или после психоэмоциональных нагрузок возникает головная боль по типу мигрени. Ребенок имеет слаборазвитые мышцы, для их укрепления можно использовать профилактор Евминова.
Постановка диагноза
Точная, своевременная диагностика синдрома Марфана способна вызвать определенные трудности, для врача это просто. Установить истину позволяет анализ родословной, исследование генов. Помимо симптомов, изучается физическое, нервно-психическое развитие. Физическое состояние изучается при помощи шкалы Стюарта.
Поставить диагноз «синдром Марфана» можно, если есть один из признаков, к примеру, вывих или частичное смещение хрусталика, аневризма аорты, аномально длинные, похожие на лапы паука, пальцы (арахнодактилия), деформация любой части грудной клетки, искривление позвоночника.
Но этого маловато, чтобы диагностировать болезнь Марфана, потребуется еще два фактора: близорукость, пролапс в области митрального клапана, подвижность суставов умеренной степени выраженности, аномально высокий рост, плоскостопие, «растяжки» на коже, пневмоторакс.
В 90% случаев диагностировать синдром Марфана удается с первого раза, при остальных 10% могут возникнуть определенные трудности. В подобной ситуации обследованию подлежит максимальное число родственников. В обязательном порядке требуется консультация генетика, офтальмологическое, кардиологическое обследование, исследование эхокардиографии. Дополнить представление врача о синдроме Марфана может рентген, измеряются индексы длины пальцев. Они вычисляются на основании рентгенологического снимка, если есть синдром Марфана, индекс будет удлинен. Определяется он на соотношении длины к толщине второй пястной кости.
Выявить характер, тяжесть заболевания сердца позволяет установить эхокардиография, ЭКГ, холтеровское мониторирование. Оценить со