Синдром Сегавы — Журнал неврологии и психиатрии им. С.С. Корсакова — 2019-04
ДОФА-зависимая дистония (синдром Сегавы) — это прогрессирующее наследственное заболевание, отличающееся полиморфными клиническими проявлениями. Синдром Сегавы, как правило, манифестирует в детском возрасте и клинически в 2—4 раза чаще выявляется у лиц женского пола. Заболевание впервые было описано в 1971 г. как наследственная прогрессирующая болезнь базальных ганглиев с выраженными дневными флюктуациями (англ.: hereditary progressive basal ganglia disease with marked diurnal fluctuation) японским неврологом Masaya Segawa на основании клинического наблюдения за двумя пациентками, каждая из которых имела дистоническую установку стоп в сочетании со спастичностью в ногах [1]. Впоследствии M. Segawa и соавт. обозначили болезнь как наследственную прогрессирующую дистонию с выраженными дневными флюктуациями (англ.: hereditary progressive dystonia with marked diurnal fluctuation) [2].
Для заболевания характерно наличие дистонии прежде всего в стопах, изменение симптомов в течение дня и их уменьшение или исчезновение на фоне приема низких доз препаратов леводопы [1—3], что позволяет дифференцировать данную патологию от других наследственных дистоний. Заболевание относится к орфанным, поэтому каждое клиническое наблюдение представляет интерес.
Клиническое наблюдение. Пациентка Т., 19 лет, поступила в Клинику нервных болезней (КНБ) им. А.Я. Кожевникова Первого Московского государственного медицинского университета им. И.М. Сеченова в апреле 2017 г. с жалобами на слабость в ногах, болезненные судороги икроножных мышц, ноющие боли в поясничном отделе позвоночника, «подворачивание стоп», тремор пальцев рук. Из анамнеза известно, что пациента родилась от здоровых родителей. Беременность протекала без патологии, роды в срок, росла и развивалась до 4 лет нормально, не отставая от сверстников. Заболела, по словам родителей, с 4 лет, когда впервые появились трудности при ходьбе — шаткость, частые падения, «ходьба на мысочках», а также слабость в ногах, дистоническая установка правой стопы («подворачивание»). Педиатры установли диагноз торсионной дистонии, но препараты L-ДОФА не были назначены. В 12 лет к симптоматике присоединились периодически возникающие миоклонии в руках и ногах, которые появлялись в любое время суток, однако затем быстро регрессировали. Пациентка отмечала ухудшение симптоматики (дистония стоп) в вечернее время. После обследования в Российской детской клинической больнице (Санкт-Петербург) больной было назначено лечение следующими лекарственными препаратами: наком (31,25 мг/сут), баклофен, беллатаминал. Миоклонии на фоне лечения регрессировали полностью и в дальнейшем возникали крайне редко и кратковременно. Дистония исчезла не полностью, достаточно часто возникала дистоническая установка стоп, преимущественно правой стопы. В последующем ежегодно проводилась курсовая терапия в стационарах без изменения терапии и существенной динамики в состоянии. В связи с тем что в феврале 2017 г. появились боли в поясничном отделе позвоночника и тазобедренных суставах, была несколько раз госпитализирована в различные больницы с диагнозом коксартроза, проводилась терапия различными комбинациями нестероидных противовоспалительных препаратов, сосудистых препаратов и витаминных комплексов с непостоянным эффектом. Базовая терапия по-прежнему включала наком (в прежней дозе), баклофен, беллатаминал, причем на этом фоне дистония стоп и нарушения походки беспокоили крайне редко. При отмене накома вновь возникала дистония стоп, более выраженная в правой стопе. Из-за сохраняющихся постоянных болей в поясничном отделе позвоночника и тремора в пальцах рук в апреле 2017 г. была госпитализирована в КНБ им. А.Я. Кожевникова Первого Московского государственного медицинского университета им. И.М. Сеченова, где был уставлен предварительный диагноз: дофа-чувствительная дистония (синдром Сегавы).
Из семейного анамнеза известно, что старшая сестра пациентки также страдает от дистонической установки стоп, однако особенности клинической картины и возраст начала заболевания неизвестны. Сестра отказалась от лечения, несмотря на то что дистония привела у нее к деформации стоп, что потребовало хирургического вмешательства. В последнее время получает наком, но дозировка и эффект неизвестны.
Больная правильного телосложения, умеренного питания. Кожные покровы обычной окраски. Костных аномалий нет. Тоны сердца приглушены, ритмичные. Артериальное давление — 120—130/70—80 мм рт.ст. В легких дыхание везикулярное, хрипов нет. Живот при пальпации мягкий, безболезненный. Симптом Пастернацкого отрицательный с обеих сторон.
Неврологический статус: больная в сознании, ориентирована в месте, времени, собственной личности. Когнитивных нарушений нет. Эмоционально лабильна. Менингеальных знаков нет. Черепная иннервация без патологии.
Двигательная сфера: мышечная сила в проксимальных отделах нижних конечностей снижена до 4 баллов, сила аксиальной мускулатуры слева достаточная, справа снижена до 4 баллов. Активные и пассивные движения в руках не ограничены. Тонус мышц конечностей не изменен. Сухожильные рефлексы живые, симметричные. Патологические рефлексы не вызываются. Динамические координаторные пробы (пальценосовая, пяточно-коленная) выполняет удовлетворительно. Постуральный тремор пальцев рук. В пробе Ромберга устойчива. Поверхностная и глубокая чувствительность на руках и ногах сохранена. Тазовые функции не нарушены.
Дистонические движения в стопах появляются при отмене накома. При отмене накома более чем на 2 дня у больной, помимо дистонической установки стоп, в большей степени левой, развилось значительное повышение мышечного тонуса в ногах, причем в левой ноге до уровня спастичности (см. рисунок). Развитие дистонической установки стоп на фоне отмены накома. При возобновлении терапии в течение 2 дней произошла нормализация неврологического статуса.
Лабораторные исследования: общий анализ крови, общий анализ мочи, биохимический анализ крови — в пределах нормы. Электрокардиограмма: синусовый ритм, частота сердечных сокращений — 64 уд/мин.
Магнитно-резонансная томография пояснично-крестцового отдела позвоночника: картина дегенеративно-дистрофических изменений тел позвонков, периневральная киста (менингоцеле) на уровне S2. Секвестированная грыжа диска на уровне L5—S1. Электронейромиография: скорость проведения возбуждения в дистальных и проксимальных отделах моторных волокон, скорость распространения возбуждения по сенсорным волокнам, параметры амплитуд моторных и сенсорных ответов нервов рук и ног в пределах нормы.
При выписке пациентке предложено продолжить прием низких доз препаратов леводопы (наком 62,5 мг/сут) и дальнейшее динамическое наблюдение у невролога.
Пациентке рекомендовано проведение молекулярно-генетического исследования для подтверждения диагноза синдрома Сегавы. После консультации с врачом-генетиком было принято решение начать диагностику с поиска мутаций в гене GCH1, различные мутации в котором ответственны за абсолютное большинство описанных в мире случаев синдрома Сегавы. Исследование кодирующей последовательности и прилежащих интронных областей гена выявило у пациентки ранее не описанный патогенный вариант с.382G>T (p.E128*) в гетерозиготном состоянии. Согласно заключению генетиков, диагноз был молекулярно-генетическими методами подтвержден.
Синдром Сегавы считается орфанным наследственным заболеванием, распространенность которого по всему миру, по одним данным, составляет 1 человек на 1 млн населения [4], а по расчетам Т. Nygaard — не более 1 человека на 2 млн населения [5]. Однако стоит учитывать, что экстремально низкая частота встречаемости данной патологии в популяции может быть связана как со сложностями диагностики, так и с постановкой неверных диагнозов пациентам, у которых дистония прогрессирует с детского возраста [4, 6]. Наследование данной патологии преимущественно аутосомно-доминантное [7—11]. Существует также аутосомно-рецессивный вариант синдрома Сегавы, связанный с мутацией фермента тирозингидроксилазы [12—15]. Вместе с тем большинство случаев ДОФА-чувствительной дистонии вызвано мутациями в гене
Заболевание манифестирует в раннем детском возрасте, средний возраст появления первых симптомов болезни составляет 6 лет [9, 10], при этом до момента дебюта болезни большинство детей развиваются нормально [11]. Клиническая картина синдрома, помимо дистонии, складывается из достаточно большого спектра неврологических синдромов: синдром паркинсонизма, синдром координаторных нарушений (в первую очередь нарушения походки), гиперрефлексия и спастичность [34—39], что порой затрудняет своевременную диагностику заболевания [40]. Однако классическими проявлениями этой наследственной патологии принято считать дистонию стоп, при этом дистония вначале охватывает одну конечность, затем — другую, а в дальнейшем распространяется и на верхние конечности. При начале заболевания в детском возрасте обычно замедляется рост, в результате чего к подростковому возрасту пациент имеет рост на два стандартных отклонения ниже нормы [7, 36]. Интеллект и психическое развитие, как правило, не страдают [7, 11]. Некоторые авторы отмечают коморбидность синдрома Сегавы с развивающимися впоследствии психоэмоциональными нарушениями (тревожные расстройства, депрессия, проблемы со сном и т. д.), однако их наличие не считается характерным [38, 39].
Из патогномоничных особенностей клинической картины стоит отметить наличие колебаний симптомов в течение дня (дневные флюктуации), т. е. пациенты отмечают ухудшение симптоматики в вечернее время и улучшение после ночного сна [39]. Снижение выраженности двигательных симптомов в ответ на прием низких доз препаратов леводопы также имеет высокое значение для постановки диагноза ДОФА-чувствительной дистонии [11, 34]. Терапию начинают с дозировки, которая не должна превышать 50—200 мг/сут, при этом эффект от лечения появляется в течение нескольких дней, реже недель [35, 37]. Примечательно, что побочные эффекты, часто развивающиеся при приеме препаратов леводопы, у данной категории пациентов проявляются крайне редко [39].
Характерным для болезни Сегавы является изменение выраженности определенной неврологической симптоматики по мере взросления пациента [2, 7, 8]. Так, при начале заболевания в раннем детском возрасте велика вероятность перехода дистонии, которая охватывает вначале одну конечность, в генерализованную форму дистонии к подростковому возрасту. В целом, как правило, к 30-летнему возрасту симптоматика перестает прогрессировать и стабилизируется. Выраженность «дневных флюктуаций» в свою очередь тоже имеет тенденцию к уменьшению по мере взросления больного, а к среднему возрасту они могут быть клинически маловыраженными, в то время как тремор, редко появляющийся у пациентов раньше подросткового возраста, имеет склонность к быстрому прогрессированию, охватывая все конечности и туловище и стабилизируясь, как правило, не раньше 30 лет [7, 9, 11].
Среди других особенностей синдрома стоит отметить различия в клинической картине в зависимости от возраста, в котором заболевание манифестировало [7, 9, 13]. Так, для пациентов, у которых синдром Сегавы начал прогрессировать с детского возраста, более характерна дистония с выраженными дневными колебаниями симптомов, в то время как у больных с началом заболевания в подростковом возрасте и старше чаще отмечено первоначальное появление тремора с отсутствием дистонии и дневных флюктуаций [7, 9].
Таким образом, большое количество особенностей, присущих синдрому Сегавы, позволяет выделять его в самостоятельную нозологическую единицу.
Ранее синдром Сегавы считался дофа-зависимым вариантом торсионной дистонии [40], однако, согласно современным представлениям о наследственных формах дистоний, которые основываются на лучшем в данный момент понимании генетических основ, патогенетических механизмов и клинической картины, характерных для этой группы заболеваний, не совсем корректно относить синдром Сегавы к варианту первичной торсионной дистонии. Большинство авторов сейчас относят синдром Сегавы к группе так называемых синдромов дистония-плюс, к которой также относят дистонию-паркинсонизм со стремительным развитием и миоклонус-дистонию [38]. Основным отличием группы синдромов дистония-плюс от обширной группы первичных торсионных дистоний или просто первичных дистоний, у которых основным и единственным (исключение составляет тремор конечностей и/или головы) клиническим проявлением является локальная или генерализованная дистония, считается наличие дополнительных симптомов или симптомокомплексов. То есть присутствие у больного каких-либо дополнительных нарушений в виде изменения мышечного тонуса или силы, наличие синдрома паркинсонизма, а также других высокоспецифичных особенностей клинического течения, к примеру положительного эффекта от приема низких доз препаратов леводопы, позволяют исключить диагноз «первичная дистония». Таким образом, наиболее правильно на данный момент классифицировать синдром Сегавы как одно из заболеваний группы «дистония-плюс синдромы».
Помимо дифференциальной диагностики синдрома Сегавы с первичными дистониями, необходимо уметь отличать его еще от ряда патологий. Наиболее часто заболевание следует дифференцировать с наследственной спастической параплегией или детским церебральным параличом, так как неврологическая симптоматика у значительного числа пациентов с диагнозом детского церебрального паралича может быть неправильно расценена в раннем возрасте. Известно, что некоторые дети из этой группы на самом деле могут иметь синдром Сегавы [6, 7, 9]. Среди других важных заболеваний, которые обязательно должны быть включены в дифференциально-диагностический поиск при подозрении на синдром Сегавы, следует упомянуть ювенильный паркинсонизм, который очень часто в начале проявляется дистонией нижних конечностей, и болезни детского возраста, связанные с нарушением метаболизма птеридина, при которых также отмечается дистония, однако характерны психомоторные нарушения и выраженная гипотония [38].
Несомненно, что основным диагностическим исследованием, позволяющим исключить синдром Сегавы или подтвердить наличие другого варианта дистонии, является молекулярно-генетический анализ [6, 7, 13]. Учитывая, что прогноз при синдроме Сегавы зависит от возраста начала лечения препаратами леводопы и является очень хорошим при высокой комплаентности к терапии, несвоевременная постановка диагноза может оказать драматический эффект на качество жизни пациента [4].
Таким образом, учитывая фундаментальные изменения, случившиеся за последние десятилетия в понимании того, что представляет собой дофа-чувствительная дистония, или синдром Сегавы, и в том, какие подходы могут быть использованы для лечения данной патологии, следующим этапом, вероятнее всего, будет внедрение молекулярно-генетических технологий, воздействующих избирательно на гены, в которых произошла мутация, что позволит изменять течение заболевания или препятствовать его возникновению [38].
Авторы заявляют об отсутствии конфликта интересов.
Сведения об авторах
Муранова Александра Валерьевна — клинический ординатор Клиники нервных болезней и нейрохирургии, ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России (Сеченовский Университет), Москва, Россия; e-mail :[email protected]
Строков Игорь Алексеевич — к.м.н., доцент кафедры нервных болезней и нейрохирургии ФГБОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России (Сеченовский Университет), Москва, Россия; e-mail: [email protected]; https://orcid.org/0000-0001-6950-7166
Казанцев Константин Юрьевич — врач Клиники нервных болезней и нейрохирургии, ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России (Сеченовский Университет), Москва, Россия; e-mail: [email protected]
Воскресенская Ольга Николаевна — д.м.н., проф. кафедры нервных болезней и нейрохирургии ФГБОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России (Сеченовский Университет), Москва, Россия; e-mail: [email protected]
*e-mail: [email protected]
Болезнь Сегавы | SAPHRIS (asenapine)

Одна из форм торсионной дистонии. Эта врожденная медленно прогрессирующая дистония, сочетающаяся с признаками паркинсонизма, клинически манифестирует у детей до 10 лет с локальной дистонией, которая в течение нескольких лет распространяется на другие части тела. Симптомы меняются в течение дня и уменьшаются на фоне приема низких доз препаратов леводопы.
Клинически ДЗД характеризуется ригидно-гипокинетическим синдромом: повышенным пластическим тонусом, различным в отдельных мышечных группах, что приводит к патологическим установкам позы. Заболевание дебютирует в возрасте до 3 лет. Вначале гиперкинезы или дистонические позы, нарастающие при произвольных движениях, возникают в одной или нескольких конечностях. Появляется медлительность при самообслуживании. Постепенно дистония распространяется на другие части тела по принципу буквы «N»: появляется в одной ноге, затем поражает руку с той же стороны, затем противоположную ногу и противоположную руку.
В первые 2 года течения поражаются две конечности, а «тетрадистония» развивается через 4 года — 5 лет. Нижние конечности страдают сильнее верхних, характер поражения асимметричен, даже на ранних стадиях. Торсионный компонент выражен умеренно. По мере течения заболевания нарастает ригидность мышц, спастический гипертонус. Пробу Ромберга дети с ДЗД выполняют с легкой неустойчивостью, несколько нарушена координация в конечностях. Сухожильные рефлексы чаще повышены, в некоторых случаях отмечаются клонусы стоп. Характерна флюктуация перечисленных симптомов – их разная выраженность в разное время суток: максимальная их выраженность к вечеру и уменьшение симптомов после сна. Назначение леводопы приводит к значительному улучшению походки в течение недели, уменьшению дистонических поз и гиперкинезов в течение 6 недель. Далее отмеченные выше явления нарастают: дети перестают ходить, могут немного ползать, затем наступает полная обездвиженность; нарастают миогенные контрактуры, стопы и кисти в постоянной патологической позе, которую исправить не удается. Появляются деформации грудной клетки, позвоночника. Дети начинают резко отставать в весе и росте, значительно уменьшается мышечная масса. Речь исчезает, часто нарушается глотание.
Дифференциальный диагноз проводят с ювенильным паркинсонизмом, болезнью Галлервордена-Шпатца, ювенильной хореей Гентингтона и болезнью Вильсона, ДЦП (спастической диплегией), спиноцеребеллярными атрофиями, миопатией, торсионной дистонией, тиками.
В случаях с дневным колебанием необходимо назначать леводопу в ежедневной дозе 10-25 мг/кг, положительный эффект подтверждает диагноз. Клинические симптомы полностью исчезают через 2-4 сут после начала терапии. Средняя доза составляет 375 мг леводопы и 37,5 мг карбидопы. Лечение можно продолжать в течение многих лет.
Вальпроаты, карбамазепин, бензодиазепины и барбитураты неэффективны; антихолинестеразные препараты ухудшают состояние. В случае ДЗД с низким уровнем серотонина в крови антидепрессанты утяжеляют дистонию.
Синдром Сегавы (реферат) — Журнал неврологии и психиатрии им. С.С. Корсакова — 2019-04
- Журналы
- Журнал неврологии и психиатрии им. С.С. Корсакова
- # 4, 2019
- Синдром Сегавы
- А. В. Муранова
ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова», Минздрава России, Москва, Россия - И. А. Строков
ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова», Минздрава России, Москва, Россия - К. Ю. Казанцев
ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова», Минздрава России, Москва, Россия - О. Н. Воскресенская
ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова», Минздрава России, Москва, Россия
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2019;119(4): 55-59
Просмотрено: 2056 Скачано: 234
Представлен клинический случай синдрома Сегавы. Обсуждаются клинические особенности заболевания, генетическая детерминированность, специфика лечения. Особое внимание уделяется дифференциальной диагностике. Ключевые слова:- синдром Сегавы
- наследственные дистонии
- торсионная дистония
- дофа-чувствительная дистония
КАК ЦИТИРОВАТЬ:
Муранова А.В., Строков И.А., Казанцев К.Ю., Воскресенская О.Н. Синдром Сегавы. Журнал неврологии и психиатрии им. С.С. Корсакова. 2019;119(4):55-59. https://doi.org/10.17116/jnevro201911904155Список литературы:
- Segawa M, Ohmi K, Itoh SA, Hayakawa H. Childhood basal ganglia disease with marked response to L-Dopa: hereditary progressive basal ganglia disease with marked diurnal fluctuation. Shinryo (Tokyo). 1971;24:667-672. (In Japanese).
- Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal fluctuation. In: Advances in neurology. Eldridge R., Fahn S. (Eds.). New York: Raven Press; 1976. https://doi.org/10.1159/000425356
- Allen N, Knopp W. Hereditary parkinsonism-dystonia with sustained control by L-DOPA and anticholinergic medication. Adv Neurol. 1976;14:201-213.
- Genetics Home References. Dopa-responsive dystonia U.S. National Library of Medicine. https://ghr.nlm.nih.gov/condition/dopa-responsive-dystonia
- Nygaard TG. Dopa-responsive dystonia. Delineation of the clinical syndrome and clues to pathogenesis. Adv Neurol. 1993;60:577-585.
- Nygaard TG, Waran SP, Levine RA, Naini AB, Chutorian AM. Dopa-responsive dystonia simulating cerebral palsy. Pediatr Neurol. 1994;11(3):236-240. https://doi.org/10.1016/0887-8994(94)90109-0
- Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol. 2003;54(suppl 6):32-45.
- Ichinose H, Ohye T, Takahashi SN, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, Fujita K, Negates T. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nature Genetics. 1994;8:236-242.
- Segawa M. Autosomal dominant GTP cyclohydrolase I (AD GCH 1) deficiency (Segawa disease, dystonia 5; DYT 5). Chang Gung Med J. 2009;32:1-11.
- Краснов М.Ю., Тимербаева С.Л., Иллариошкин С.Н. Генетика наследственных форм дистонии. Анналы неврологии. 2013;7:2. www.annaly-nevrologii.ru
- Бобылова М.Ю., Михайлова С.В., Гринио Л.П. Дофа-зависимая дистония (болезнь Сегавы). Журнал неврологии и психиатрии им. С.С. Корсакова. 2009;109(8):73-76
- Thony B, Blau N. Mutations in the Bh5-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Human Mutation. 2006;27(9):870-878. https://doi.org/10.1002/humu.20366
- Lee W-W, Jeon BS. Clinical spectrum of dopa-responsive dystonia and related disorders. Curr Neurol Neurosci Rep. 2014;14(7):461. https://doi.org/10.1007/s11910-014-0461-9
- Cai Chunyou, Shi W, Zeng Z, Zhang M, Ling C, Chen L, Cai Chunquan, Zhang B, Li W-D. GTP cyclohydrolase I and tyrosine hydroxylase gene mutations in familial and sporadic dopa-responsive dystonia patients. PLoS One. 2013;8(6):e65215. https://doi.org/10.1371/journal.pone.0065215
- Fletcher NA, Holt IJ, Harding AE, Nygaard TG, Mallet J, Marsden CD. Tyrosine hydroxylase and levodopa responsive dystonia. J Neurol Neurosurg Psychiat. 1989;52:112-114. https://doi.org/10.1136/jnnp.52.1.112
- Wider C, Melquist S, Hauf M, Solida A, Cobb SA, Kachergus JM, Gass J, Coon KD, Baker M, Cannon A, Stephan DA, Schorderet DF, Ghika J, Burkhard PR, Kapatos G, Hutton M, Farrer MJ, Wszolek ZK, Vingerhoets FJG. Study of a Swiss dopa-responsive dystonia family with a deletion in GCh2: redefining DYT14 as DYT5. Neurology. 2008;70:1377-1383. https://doi.org/10.1212/01.wnl.0000275527.35752.c5
- Grotzsch H, Pizzolato G-P, Ghika J, Schorderet D, Vingerhoets FJ, Landis T, Burkhard PR. Neuropathology of a case of dopa-responsive dystonia associated with a new genetic locus, DYT14. Neurology. 2000;58:1839-1842. https://doi.org/10.1212/wnl.58.12.1839
- Ichinose H, Suzuki T, Inagaki H, Ohye T, Nagatsu T. Molecular genetics of dopa-responsive dystonia. Biol Chem. 1999;380(12):1355-1364. https://doi.org/10.1515/bc.1999.175
- Ichinose H, Inagaki H, Suzuki T, Ohye T, Nagatsu T. Molecular mechanisms of hereditary progressive dystonia with marked diurnal fluctuation, Segawa’s disease. Brain and Development. 2000;22(suppl 1):107-110. https://doi.org/10.1016/s0387-7604(00)00136-4
- Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K, Mohrmann K, Lohnau T, Raymond D, Vieregge P, Nygaard T, Ozelius LJ, Bressman SB, Klein C. High mutation rate in dopa-responsive dystonia: detection with comprehensive GCh2 screening. Neurology. 2005;64:908-911. https://doi.org/10.1212/01.wnl.0000152839.50258.a2
- Furukawa Y. Genetics and biochemistry of dopa-responsive dystonia: significance of striatal tyrosine hydroxylase protein loss. Adv Neurol. 2003;91:401-410.
- Zirn B, Steinberger D, Troidl C, Brockmann K, von der Hagen M, Feiner C, Henke L, Muller U. Frequency of GCh2 deletions in dopa-responsive dystonia. J Neurol Neurosurg Psychiat. 2008;79:183-186. https://doi.org/10.1136/jnnp.2007.128413
- Cao L, Zheng L, Tang W-G, Xiao Q, Zhang T, Tang H-D, He S-B, Wang Xi-Jin, Ding J-Q, Chen S-D. Four novel mutations in the GCh2 gene of Chinese patients with dopa-responsive dystonia. Movement Disorders. 2010;25(6):755-783. https://doi.org/10.1002/mds.22646
- Liu X, Zhang S-S, Fang D-F, Ma M-Y, Guo X-Y, Yang Y, Shang H-F. GCh2 Mutation and clinical study of Chinese patients with dopa-responsive dystonia. Movement Disorders. 2010;25(4):447-451. https://doi.org/10.1002/mds.22976
- Tamaru Y, Hirano M, Ito H, Kawamura J, Matsumoto S, Imai T, Ueno S. Clinical similarities of hereditary progressive/dopa responsive dystonia caused by different types of mutations in the GTP cyclohydrolase I gene. J Neurol Neurosurg Psychiat. 1998;64(4):469-473. https://doi.org/10.1002/ana.410400517
- Scola RH, Carducci C, Amaral VG, Lorenzoni PJ, Teive HA, Giovanniello T, Werneck LC. A novel missense mutation pattern of the GCh2 gene in dopa-responsive dystonia. Arq Neurosiquiatr. 2007;65:1224-1227. https://doi.org/10.1590/s0004-282×2007000700026
- Robinson R, McCarthy GT, Bandmann O, Dobbie M, Surtees R, Wood NW. GTP cyclohydrolase deficiency; intrafamilial variation in clinical phenotype, including levodopa responsiveness. J Neurol Neurosurg Psychiat. 1999;66(1):86-89. https://doi.org/10.1136/jnnp.66.1.86
- Furukawa Y, Kish SJ. Dopa-responsive dystonia: recent advances and remaining issues to be addressed. Mov Disord. 1999;14(5):709-715. https://doi.org/10.1002/1531-8257(199909)14:5<709::AID-MDS1001>3.0.CO;2-T
- Steinberger D, Weber Y, Korinthenberg, R, Deuschl G, Benecke R, Martinius J, Muller U. High penetrance and pronounced variation in expressivity of GCh2 mutations in five families with dopa-responsive dystonia. Ann Neurol. 1998;43:634-639. https://doi.org/10.1002/ana.410430512
- Furukawa Y, Lang AE, Trugman JM, Bird TD, Hunter A, Sadeh M. Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology. 1998;50(4):1015-1020. https://doi.org/10.1212/wnl.50.4.1015
- Müller U, Steinberger D, Topka H. Mutations of GCh2 in Dopa-responsive dystonia. J Neural Transm. 2002;109:321-328. https://doi.org/10.1007/s007020200026
- Nygaard TG, Snow BJ, Fahn S, Calne DB. Dopa-responsive dystonia: clinical characteristics and definition. In: Segawa M. (ed). Hereditary progressive dystonia with marked diurnal fluctuation. Parthenon Publishing. 1993.
- Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabro B, Landrieu P, Nguyen K, Ponsot G, Abada M, Doummar D, Damier P, Gil R, Thobois S, Ward AJ, Hutchinson M, Toutain A, Picard F, Camuzat A, Fedirko E, San C, Bouteiller D, LeGuern E, Durr A, Vidailhet M, Brice A, French Dystonia Network. Exhaustive analysis of Bh5 and dopamine biosynthesis genes in patients with dopa-responsive dystonia. Brain. 2009;132:1753-1763. https://doi.org/10.1093/brain/awp084
- Chaila EC, McCabe DJ, Delanty N, Costello DJ, Murphy RP. Broadening the phenotype of childhood-onset dopa-responsive dystonia. Arch Neurol. 2006;63(8):1185-1188. https://doi.org/10.1001/archneur.63.8.1185
- Nygaard TG, Trugman JM, de Yebenes JG, Fahn S. Dopa-responsive dystonia: the spectrum of clinical manifestations in a large North American family. Neurology. 1990;66:66-69. https://doi.org/10.1212/wnl.40.1.66
- van Egmond ME, Kuiper A, Eggink H, Sinke RJ, Brouwer OF, Verschuuren-Bemelmans CC, Sival DA, Tijssen MAJ, de Koning TJ. Dystonia in children and adolescents: a systematic review and a new diagnostic algorithm. J Neurol, Neurosurg, Psychiat. 2015;86(7):774-781. https://doi.org/10.1136/jnnp-2014-309106
- Hahn H, Trant MR, Brownstein MJ, Harper RA, Milstien S, Butler IJ. Neurologic and psychiatric manifestations in a family with a mutation in exon 2 of the guanosine triphosphate-cyclohydrolase gene. Arch Neurol. 2001;58:749-755. https://doi.org/10.1001/archneur.58.5.749
- Phukan J, Albanese A, Gasser T, Warner T. Primary dystonia and dystonia-plus syndromes: clinical characteristics, diagnosis, and pathogenesis. Lancet Neurol. 2011;10:1074-1085. https://doi.org/10.1016/s1474-4422(11)70232-0
- Steinberger D, Korinthenberg R, Topka H, Berghauser M, Wedde R, Muller U. Dopa-responsive dystonia: mutation analysis of GCh2 and analysis of therapeutic doses of L-dopa. Neurology. 2000;55:1735-1737. https://doi.org/10.1212/wnl.55.11.1735
- Вельтищев Ю.Е., Темин П.А. Наследственные болезни нервной системы. Руководство для врачей. 1998.
7 лет и 4 дня или опыт лечения болезни Сегавы в ММЦ «СОГАЗ»
7 лет и 4 дня или опыт лечения болезни Сегавы в ММЦ «СОГАЗ»
В октябре состоялся VII КсеоФорум, площадкой для которого, в этот раз, стал гостеприимный г.Баку (Азербайджан), встретивший участников конференции со свойственным радушием и теплом.Представители России, а также стран ближнего и дальнего зарубежья, в седьмой раз приняли участие в конференции, посвященной проблемам изучения и лечения заболеваний нервной системы с применением Ботулинического токсина типа А (Ксеомин).
Ведущий врач-невролог ММЦ «СОГАЗ» Шахметова О.А. выступила с двумя докладами. Первый был посвящен использованию ботулотоксина при лечении краниомандибулярной дисфункции, осложненной прозопалгией мышечного генеза. В работе был представлен опыт комплексного лечения лицевой боли на фоне дисфункции височно-нижнечелюстного сустава. Лечение состояло из двух этапов: окклюзионной терапии (ношение жестких капп) и ботулинотерапии жевательной группы мышц.
Во втором выступлении был представлен доклад на тему: «Болезнь Сегавы. Клинический случай» — об успешном опыте лечения болезни Сегавы в ММЦ «СОГАЗ», что вызвало живой интерес коллег-участников конференции.
В июле 2016г. в ММЦ «СОГАЗ» обратилась семья 12-ти летней пациентки, с признаками болезни Сегавы. В попытках узнать причину мучительного состояния, в котором пребывал их ребенок в течение 7 лет, семья девочки обращалась во многие медицинские учреждения России и зарубежья. Однако, правильно диагностировать заболевание врачи не смогли.
Дело в том, что болезнь Сегавы (ДОФА -чувствительная дистония) — это довольно редкое генетическое заболевание, встречается один случай на миллион. Впервые болезнь была описана японским неврологом M.Segawa в 1976г. Чаще всего заболевание проявляется в возрасте 5-8 лет у девочек и характеризуется нарушением походки и дистонией. Со временем могут проявляться признаки паркинсонизма. Однако, при своевременном и корректном лечении прогноз для пациентов, страдающих болезнью Сегавы самый положительный.
На основании клинической картины, доктором Шахметовой О.А было диагностировано данное заболевание и назначена заместительная терапия дофамином в микродозах. По результатам которой, уже на четвертые сутки, девочка почувствовала качественное улучшение состояния. Симптомы исчезли и пациентка была выписана домой с назначением заместительной терапии.
Сейчас девочка чувствует себя хорошо, посещает школу и ведет обычную жизнь подростка.
Этот случай еще раз подтверждает, что в нашем центре работают специалисты с мировой практикой лечения различных заболеваний, в том числе и довольно редких.
24 Октября 2016
Болезнь Сегавы (ДОФА-чувствительная торсионная дистония, DYT4)
Болезнь Сегавы (ДОФА-чувствительная торсионная дистония, DYT4)
Болезнь Сегавы (ДОФА-чувствительная дистония) относится к торсионным дистониям и наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Ген находится на длинном плече 14-й хромосомы . Заболевание начинается в детском возрасте. Дистония сочетается с гипокинезией и ригидностью . Это заболевание характеризуется дистонией в сочетании с паркинсонизмом — причем у 70% больных отмечается выраженная вариабельность выраженности клинической картины в течении суток ( Nygaard et al, 1991 , Nygaard et al, 1993 ). Женщины болеют примерно в 4 раза чаще мужчин. Возраст начала заболевания и выраженность симптомов паркинсонизма сильно варьирует — у части больных отмечена изолированная дистония, в некоторых случаях заболевание манифестирует как фокальная дистония мышц гортани или изолированное нарушение позы стоп ( Nygaard et al, 1988 , Ichinose et al 1988 , Иванова-Смоленская И.А. и др., 1996 ).
Заболевание является редким — частота его встречаемости не превышает 1 случая на миллион.
ДОФА-зависимая дистония — аутосомно-доминантное заболевание, пенетрантность которого оценивается в 30%. Однако учет атипичных и стертых проявлений заболевания значительно увеличивает пентрантность и не исключено, что она может достигать 100% ( Nygaard et al, 1990 , Steinberger et al, 1998 ).
Причиной заболевания являются мутации в гене ГТФ циклогидролазы 1 — первого фермента цикла биосинтеза тетрагидробиоптерина , кофактора биосинтеза ароматических аминокислот и в том числе L-ДОФА ( рисунок 7 ) ( Nygaard et al, 1993 , Ichinose, 1994 ).
Лечение: см. Торсионная дистония: лечение .
Ссылки:
Дофа-зависимая дистония (болезнь Сегавы). Дистония у детей
Описание клинической картины и классификация форм Дофа-зависимой дистонии. Изучение причин, механизмов патогенеза, принципов молекулярной диагностики и алгоритма лечения заболевания. Рассмотрение генетики дистонии и процесса биосинтеза катехоламинов.
Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
HTML-версии работы пока нет.
Cкачать архив работы можно перейдя по ссылке, которая находятся ниже.
Подобные документы
Причины возникновения вегетососудистой дистонии. Симптомы заболевания и особенности его проявления. Лечебная программа при ВСД. Комплекс физических упражнений при повышенном или пониженном давлении. Дыхательная гимнастика при вегетативной дистонии.
реферат , добавлен 18.04.2013
Что нужно знать о заболевании: работа сосудов, проявления вегетососудистой дистонии, причины возникновения. Коррекция образа жизни — основа исцеления. Роль питания и физической активности в профилактике заболевания. Лечение вегетососудистой дистонии.
реферат , добавлен 28.12.2010
Основные симптомы и причины возникновения заболевания. Обоснование механизмов лечебного действия физических упражнений. Показания и противопоказания к назначению лечебной физкультуры. Запрещенные виды физической нагрузки при вегето-сосудистой дистонии.
реферат , добавлен 31.05.2013
Физиология вегетативной нервной системы. Возрастные периоды риска формирования вегетативных нарушений. Причины возникновения, провоцирующие факторы. Патогенез дистонии: классификация, симптомы. Дифференциальная диагностика. Принципы лечения заболевания.
презентация , добавлен 02.02.2012
Определение вегетативно-сосудистой и нейроциркулярной дистонии. Особенности их лечения. Исследование воздействия различных средств физической реабилитации на функциональное состояние больных НЦД. Комплекс упражнений. Периоды физической реабилитации.
реферат , добавлен 05.12.2009
Вегето-сосудистая дистония: этиология, патогенез, клинические проявления. Анатомия и физиология нервной системы, методы исследования. Методики водолечения при вегето-сосудистой дистонии. Массаж и самомассаж, основные методы психологической коррекции.
курсовая работа , добавлен 16.05.2012
Этиология заболевания, внутренние и внешние факторы, способствующие возникновению вегето-сосудистой дистонии. Клинические проявления и лечение заболевания. Виды физической активности, наиболее полезной людям с вегетативными и сосудистыми нарушениями.
Торсионная дистония — наследственное заболевание с поражением опорно-двигательного аппарата. На сегодняшний день выделяют 17 форм данного заболевания (табл.1). Синим шрифтом в таблице выделены формы торсионной дистонии, ДНК-диагностика которых проводится в Центре Молекулярной Генетики. По результатам лечения препаратами L-дофа некоторые формы классифицируются на ДОФА-зависимую (ригидная) и ДОФА-независимую (гиперкинетическая) формы.
Таблица 1. Формы торсионной дистонии
Форма дистонии | Название заболевания | Тип наследования | ||
Торсионная дистония | ||||
Торсонная дистония | ||||
Торсонная дистония-паркинсонизм | Х-сцепленный | |||
Торсионная дистония | ||||
ДОФА-зависимая дистония | ||||
Торсионная дистония с поздним началом | ||||
Хореоатетоз, некинезогенная дискинезия | ||||
Дистония 9 | ||||
Пароксизмальный кинезогенный хореоатетоз | ||||
Миоклоническая дистония | ||||
Дистония-паркинсонизм со стремительным развитием | ||||
Торсионная дистония | ||||
ДОФА-зависимая дистония | ||||
Миоклоническая дистония | ||||
Дистония 16 | ||||
Торсионная дистония |
Дистония 1
ДОФА-независимая (гиперкинетическая) форма с аутосомно-доминантным типом наследования обычно начинается на 1-м десятилетии жизни, характеризуется развитием вычурных «скручивающих» генерализованных гиперкинезов конечностей, туловища и шеи, резко усиливающихся при ходьбе и приводящих к тяжёлой инвалидизации больного.
Наиболее частой мутац
Дофа-чувствительная дистония (болезнь Сегавы)
Самым ярким недавним событием в изучении дистонии стало обнаружение формы генерализованной дистонии, при которой эффективна леводофа.
Болезнь начинается в детстве. Вначале страдают ноги (больные ходят на цыпочках, не сгибая колен, часто падают и подворачивают стопу), а затем дистония медленно распространяется на руки и туловище; часто присоединяются брадикинезия и ригидность. Дистония в ногах может почти отсутствовать по утрам, резко нарастая к вечеру и после физической нагрузки. При этой форме нередко ошибочно диагностируют гиперкинетическую форму детского церебрального паралича. Главный отличительный признак этой формы — поразительный положительный эффект леводофы (синтетический левовращающий изомер диоксифенилаланина — L-дофа,).
Аутосомно-доминантная форма болезни обусловлена мутацией гена GCh2, локализованного в сегменте 14q22. Этот ген кодирует ГТФ-циклогидролазу I, участвующую в синтезе биоптерина — кофактора тирозингидроксилазы, необходимого для синтеза дофамина. В результате мутации резко снижается содержание дофамина в полосатом теле.
3. Материнская ФКУ
Этиология
Появление умственной отсталости среди потомства женщин с ФКУ, не соблюдающих диету в зрелом возрасте, получило наименование материнской ФКУ.
Патогенез
Патогенез патологии мало изучен, однако предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Причем в связи с накоплением этой аминокислоты в плаценте, ее содержание в организме плода оказывается выше, чем у матери. Тем не менее, прямое токсическое действие фенилаланина точно не подтверждено.
Нарушение обмена тирозина.
Тирозин, помимо участия в синтезе белков, является предшественником гормонов надпочечников адреналина, норадреналина, медиатора дофамина, гормонов щитовидной железы тироксина и трийодтиронина, пигментов. Нарушения обмена тирозина многочисленны и называются тирозинемии.
Тирозинемии
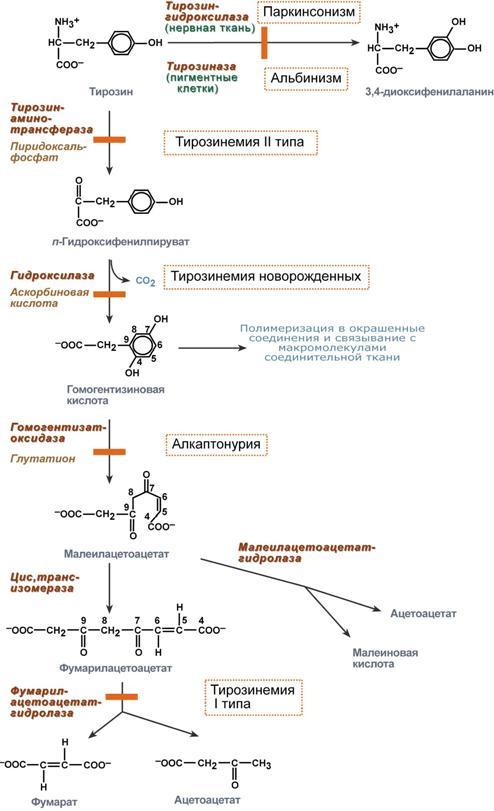
Причины возникновения тирозинемий
Тирозинемия 1 типа
Этиология
Тирозинемия типа I (гепаторенальная тирозинемия) возникает при недостаточности фумарилацетоацетат-гидролазы. При этом накапливается фумарилацетоацетат и его метаболиты, поражающие печень и почки.
Клиническая картина
Существует две формы – остраяихроническая.
Острая формасоставляет большинство случаев заболевания с началом в возрасте 2-7 мес и смертью 90% больных в возрасте 1-2 года из-за недостаточности печени.
К симптомам относится гипотрофия,рвота, «капустный запах» от тела и мочи,задержка развития, кровоточивость, диарея,мелена(дёгтеобразный стул – признак кровотечения в ЖКТ),гематурия, желтуха, анемия, периферические невропатии и параличи, кардиомиопатия, слабость мышц, дыхательные нарушения. Отмечаютгипогликемиювследствие гиперплазии островковых клеток поджелудочной железы.
При хронической формеболезнь развивается позднее, медленнее прогрессирует. Продолжительность жизни около 10 лет.
Наблюдаются гипотрофия, узелковый цирроз печениипеченочная недостаточность, множественныедефекты почечной канальцевой реабсорбции с появлением синдрома Фанкони (щелочная рН мочи, глюкозурия, протеинурия),аминоацидурия, лейкопения, тромбоцитопения.
Из-за поражения печени и почек возникают проявления рахитоподобных заболеваний (остеопороз, остеомаляция). В результате печеночной недостаточности возникают симптомы, напоминающие острую порфирию. Непостоянными признаками являютсяумственная отсталостьи неврологические изменения.
болезней органов пищеварения | Информационный центр по генетическим и редким заболеваниям (GARD) — программа NCATS
Синдром делеции 22q11.2
Синдром Aagenaes
Абеталипопротеинемия
Добавочная поджелудочная железа
Синдром микроцефалии ахалазии
Энтеропатический акродерматит
Острая жирная печень при беременности
Полиглюкозановая болезнь тела взрослых
Агенезия дорсальной части поджелудочной железы
Синдром Аль-Газали-Доннаи-Мюллера
ALG13-CDG
ALG2-CDG (CDG-Ii)
ALG6-CDG (CDG-Ic)
ALG8-CDG (CDG-Ih)
ALG9-CDG (CDG-IL)
Синдром Альперса
Дефицит антитрипсина альфа-1
Ankyloblepharon filiforme неперфорированный задний проход
Кольцевидная поджелудочная железа
Aplasia cutis congenita кишечная лимфангиэктазия
Синдром артериальной извитости
Синдром холестаза дисфункции почек артрогрипоз
Синдром искусств
Атрезия тонкой кишки
Аутоиммунная нарушение моторики желудочно-кишечного тракта
Аутоиммунный гепатит
Аутоиммунный лимфопролиферативный синдром, связанный с гаплонедостаточностью CTLA4
Аутосомно-рецессивное воспалительное заболевание кишечника с ранним началом
Синдром Аксенфельда-Ригера
B4GALT1-CDG (CDG-IId)
Синдром Баллера-Герольда
Синдром Баннаяна-Райли-Рувалькабы
Банту сидероз
Синдром Барде-Бидля
Синдром Барде-Бидля 1
Синдром Барде-Бидля 10
Синдром Барде-Бидля 11
Синдром Барде-Бидля 12
Синдром Барде-Бидля 2
Синдром голых лимфоцитов 2
Пищевод Барретта — Не редкое заболевание
Доброкачественный рецидивирующий внутрипеченочный холестаз 1
Доброкачественный рецидивирующий внутрипеченочный холестаз 2
Бифидный нос с аноректальными и почечными аномалиями или без них
Рак желчных протоков
Атрезия желчных путей
Синдром Бурхааве
Синдром Бадда-Киари
Синдром Канту
Болезнь Кароли
Синдром кошачьего глаза
Последовательность каудальной регрессии
Церебротехнический ксантоматоз
Гепатоцеллюлярная карцинома у детей
Болезнь накопления холестерилового эфира
Хроническая гранулематозная болезнь
Хроническая икота
Болезнь задержки хиломикронов
Хилезный асцит
Цитруллинемия II типа
Классический синдром Элерса-Данлоса
Синдром тренера
COG4-CDG (CDG-IIj)
Коллагенозный колит — Не редкое заболевание
Коллагеновый гастрит
Врожденный дефект синтеза желчной кислоты 1 тип
Врожденный дефект синтеза желчных кислот 2 типа
Врожденная хлоридная диарея
Врожденная диафрагмальная грыжа
Врожденные нарушения гликозилирования
Врожденная лактазная недостаточность
,00 Врожденная недостаточность сахаразы-изомальтазы
Синдром Корнелии де Ланге
Синдром Каудена
Синдром Криглера-Наджара, тип 1
Синдром Криглера-Наджара тип 2
болезнь Крона — Не редкое заболевание
Кронхайт-канадская болезнь
Куррарино триада
Кожная светочувствительность и колит, летальный
Cutis laxa, аутосомно-доминантный
Cutis laxa, аутосомно-рецессивный тип 1
Муковисцидоз
Киста Денди-Уокера с дисплазией почек-печени-поджелудочной железы
DDOST-CDG (CDG-Ir)
Глухота, дистония и церебральная гипомиелинизация
Десмопластическая мелкоклеточная опухоль
Диссеминированный перитонеальный лейомиоматоз
Синдром Доннаи-Барроу
DPM2-CDG
Синдром Дубина-Джонсона
Атрезия двенадцатиперстной кишки
Язва двенадцатиперстной кишки, вызванная гиперфункцией антральных G-клеток
Синдром Эмануэля
Эозинофильный гастроэнтерит
Атрезия пищевода
Экстрофия мочевого пузыря
Семейная каудальная дисгенезия
Семейный рак поджелудочной железы
Семейная висцеральная миопатия с наружной офтальмоплегией
Синдром Фанкони-Бикеля
Синдром Фейнгольда
Синдром Фрейзера
Синдром Фростера-Хуха
Синдром Фринса
Дефицит галактокиназы
Дефицит галактозоэпимеразы
Синдром Гарднера
Желудочно-кожный синдром
Стромальные опухоли желудочно-кишечного тракта
Гастрошизис
Остеодиспластическая геродермия
Нарушение всасывания глюкозы-галактозы
Болезнь накопления гликогена типа 1А
Болезнь накопления гликогена типа 1B
Болезнь накопления гликогена типа 3
Болезнь накопления гликогена типа 6
Карциноид бокаловидных клеток
Синдром мегаколона Гольдберга-Шпринцена
ГРАЦИЛЬНЫЙ синдром
Гемохроматоз 2 типа
Гемохроматоз 3 типа
Гемохроматоз 4 типа
Печеночная энцефалопатия
Веноокклюзионная болезнь печени
Веноокклюзионная болезнь печени с иммунодефицитом
Гепатобластома
Наследственный диффузный рак желудка
Наследственная мальабсорбция фолиевой кислоты
Наследственная непереносимость фруктозы
Наследственная геморрагическая телеангиэктазия
Наследственная геморрагическая телеангиэктазия 2 типа
Наследственная геморрагическая телеангиэктазия 3 типа
Наследственная геморрагическая телеангиэктазия 4 типа
Наследственный панкреатит
Болезнь Гиршпрунга тип d брахидактилия
Болезнь Гиршпрунга
Гипербилирубинемия преходящая семейная неонатальная
Ихтиоз, лейкоцитарные вакуоли, алопеция и склерозирующий холангит
Идиопатическая ахалазия
Синдром Имерслунда-Грасбека
Иммунодисрегуляция, полиэндокринопатия и энтеропатия Х-сцепленная
Синдром детской печеночной недостаточности 1
Инфантильная спиноцеребеллярная атаксия
Множественная атрезия кишечника
Внутрипеченочный холестаз при беременности
Атрезия Jejunal
Синдром Йохансона-Близзарда
Синдром ювенильного полипоза
Синдром Кабуки
Kernicterus
Опухоль Клацкина
Комплекс стенок конечности и тела
Дефицит LRBA
Синдром Люси-Дрисколла
Синдром Линча — Не редкое заболевание
Малакоплакия
Синдром Мэллори-Вейсса
Синдром Меккеля
Синдром гипоперистальтики кишечника Megacystis microcolon
Мегадуоденум и / или мегацистис
Болезнь Менетрие
Умственная отсталость скелетная дисплазия abducens palsy
Дефект редукции конечности микрогастрии
Синдромный микрофтальм 9
Микрофтальм с синдромом линейных дефектов кожи
Болезнь включения микроворсинок
Синдром митохондриальной нейрогастроинтестинальной энцефалопатии
MOGS-CDG (CDG-IIb)
MPI-CDG (CDG-Ib)
Синдром истощения митохондриальной ДНК, связанный с MPV17
Синдром Мюира-Торре
Множественная эндокринная неоплазия 1 типа
Множественная эндокринная неоплазия 2А типа
Множественная эндокринная неоплазия 2B типа
Синдром мультисистемной дисфункции гладких мышц
Ассоциация MURCS
Некротический энтероколит
Неонатальная адренолейкодистрофия
Неонатальный гемохроматоз
Узловая регенеративная гиперплазия
Синдром затылочного рога
Омфалоцеле синдром волчьей пасти летальный
Омфалоцеле, экстрофия клоаки, неперфорированный задний проход и комплекс дефектов позвоночника
Омфаломезентериальная киста
Синдром Паллистера-Холла
Мозаичный синдром Паллистера-Киллиана
Аденома поджелудочной железы
Рак поджелудочной железы
Синдром Пирсона
Детская болезнь Крона
Детский язвенный колит
Пенталогия Кантрелла
Синдром Пейтца-Егерса
PGM1-CDG
Синдром Пламмера Винсона
PMM2-CDG (CDG-Ia)
Поликистоз печени
Первичный билиарный холангит
Первичная кишечная лимфангиэктазия
Первичный рак печени
Первичный склерозирующий холангит
Прогрессирующий семейный внутрипеченочный холестаз 1
Прогрессирующий семейный внутрипеченочный холестаз 2 типа
Прогрессирующий семейный внутрипеченочный холестаз 3 типа
Прогрессирующий семейный внутрипеченочный холестаз-4
Псевдомиксома брюшины
Болезнь Рефсума, младенческая форма
Синдром почечного щелкунчика
Забрюшинный фиброз
Синдром Рейнольдса
RFT1-CDG (CDG-In)
Кольцевая хромосома 13
Синдром ротора
Синдром Сандифера
Синдром Сатойоши
Синдром шарфа
Склерозирующий мезентериит
Синдром коротких ребер-полидактилии тип 3
Синдром Шпринцена омфалоцеле
Синдром Швахмана-Даймонда
Синдром Симпсона-Голаби-Бемеля
Сиреномелия
Аденокарцинома тонкого кишечника
Спленогонадальные дефекты конечностей, микрогнатия
Синдром сталкера Читаята
СТАР синдром
Синдром верхней брыжеечной артерии
Синдромный микрофтальм 3 типа
Торако-абдоминальная кишечная дупликация
TMEM165-CDG (CDG-IIk)
Синдром Таунса-Брокса
Транзиторная детская печеночная недостаточность
Трихогепатоэнтерический синдром
Синдром тройного А
Трисомия 13
Трисомия 18
Тафтинговая энтеропатия
Тилоз при раке пищевода
Тирозинемия 1 типа
Ульнарно-молочный синдром
ВАКТЕРЛ ассоциация
VIPoma
Синдром Ваарденбурга тип 4
Арбузный желудок
Болезнь Уиппла
Болезнь Вильсона
Синдром Вольфа-Хиршхорна
Болезнь Вольмана
Синдром морщинистой кожи
Синдром Зеллвегера
Синдром Золлингера-Эллисона
.
Болезнь Канавана — Genetics Home Reference
Baslow MH, Guilfoyle DN. Болезнь Канавана, редкая губчатая лейкодистрофия человека с ранним началом: понимание ее генезиса и возможных клинических вмешательств. Биохимия. 2013 Апрель; 95 (4): 946-56. DOI: 10.1016 / j.biochi.2012.10.023. Epub 2012 11 ноября.
Фейгенбаум А., Мур Р., Кларк Дж., Хьюсон С., Читаят Д., Рэй П.Н., Стокли Т.Л. Болезнь Канавана: определение частоты носителей у еврейского населения ашкенази и разработка нового молекулярного диагностического теста.Am J Med Genet A. 2004, 15 января; 124A (2): 142-7.
Guo F, Bannerman P, Mills Ko E, Miers L, Xu J, Burns T, Li S, Freeman E, McDonough JA, Pleasure D. Удаление N-ацетиласпартата предотвращает лейкодистрофию в модели болезни Канавана. Энн Нейрол. 2015 Май; 77 (5): 884-8. DOI: 10.1002 / ana.24392. Epub 2015 27 марта.
Янсон К.Г., Макфи С.В., Фрэнсис Дж., Шера Д., Ассади М., Фриз А., Хур П., Хазелгроув Дж., Ван Д.Дж., Биланюк Л., Леоне П. Естественная история болезни Канавана, выявленная протоном магнитно-резонансная спектроскопия (1H-MRS) и диффузионно-взвешенная МРТ.Нейропедиатрия. 2006 Август; 37 (4): 209-21.
Мадхаварао С.Н., Арун П., Моффетт Дж. Р., Сзукс С., Сурендран С., Маталон Р., Гарберн Дж., Христова Д., Джонсон А., Цзян В., Намбудири М. А.. Дефектный катаболизм N-ацетиласпартата снижает уровень ацетата в мозге и синтез липидов миелина при болезни Канавана. Proc Natl Acad Sci U S. A. 2005 5 апреля; 102 (14): 5221-6. Epub 2005 22 марта.
Маталон Р., Михалс-Маталон К. Болезнь Канавана. 1999 16 сентября [обновлено 11 августа 2011]. В: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, редакторы.GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2017 гг. Доступно по адресу http://www.ncbi.nlm.nih.gov/books/NBK1234/
Namboodiri AM, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN. Болезнь Канавана и роль N-ацетиласпартата в синтезе миелина. Mol Cell Endocrinol. 2006 27 июня; 252 (1-2): 216-23. Epub 2006 2 мая. Обзор.
Сурендран С., Бамфорт Ф.Дж., Чан А., Тайринг С.К., Гудман С.И., Маталон Р.Легкое повышение уровня N-ацетиласпарагиновой кислоты и макроцефалия: диагностическая проблема. J Child Neurol. 2003 ноя; 18 (11): 809-12.
Сурендран С., Михалс-Маталон К., Кваст М.Дж., Тайринг С.К., Вей Дж., Эзелл Е.Л., Маталон Р. Болезнь Канавана: моногенная черта со сложным геномным взаимодействием. Mol Genet Metab. 2003 сентябрь-октябрь; 80 (1-2): 74-80. Обзор. Ошибка в: Mol Genet Metab. 2006 Март; 87 (3): 279.
Tacke U, Olbrich H, Sass JO, Fekete A, Horvath J, Ziyeh S, Kleijer WJ, Rolland MO, Fisher S, Payne S, Vargiami E, Zafeiriou D.I, Omran H.Возможные генотип-фенотипические корреляции у детей с легким течением болезни Канавана. Нейропедиатрия. 2005 август; 36 (4): 252-5.
Болезнь Джона — Американская ассоциация молочных коз
Болезнь Джона («YO-колени») — это смертельное желудочно-кишечное заболевание коз и других жвачных животных (включая крупный рогатый скот, овец, лосей, оленей и бизонов), вызываемое бактерией Mycobacterium avium subpecies paratuberculosis (MAP). Эта инфекция, также известная как паратуберкулез, заразна, что означает, что она может распространяться в вашем стаде.

Болезнь Джона
Вопросы и ответы
для владельцев коз
Национальная образовательная инициатива Джона отмечаетЭлизабет Паттон и доктору Гретхен Мэй из Департамента сельского хозяйства, торговли и защиты потребителей Висконсина и доктору Элизабет Мэннинг из Информационного центра Университета Висконсина в Мэдисоне Джона за их вклад в эту статью. Некоторые фотографии были предоставлены Информационным центром Джона, Университет Висконсин-Мэдисон, http://johnes.org.
В: Что такое болезнь Джона?
A: Болезнь Джона («Йо-колени») — это фатальное желудочно-кишечное заболевание коз и других жвачных животных (включая крупный рогатый скот, овец, лосей, оленей и бизонов), которое вызывается бактерией Mycobacterium avium subpecies paratuberculosis (MAP). .Эта инфекция, также известная как паратуберкулез, заразна, что означает, что она может распространяться в вашем стаде.
Микроорганизм MAP чаще всего передается с навозом инфицированных животных. Инфекция обычно передается от взрослых коз к детям и происходит, когда молодое животное проглатывает организм через воду, молоко или корм, загрязненный навозом инфицированных животных. Большинство владельцев застают врасплох, когда диагностируют инфекцию, и слишком поздно узнают, что инфекция охватила несколько животных в стаде.
Из-за отсутствия тестирования и отчетности неизвестно, насколько широко распространен
Болезнь Джона встречается у коз в Соединенных Штатах. Однако инфекция была подтверждена во многих козьих стадах по всей стране — молочных, мясных, традиционных и других пород — и это также проблема в большинстве других стран, разводящих коз. Издержки этой инфекции варьируются от экономических потерь — из-за сокращения производства и увеличения выбраковки мясных и молочных животных — до эмоциональных потерь — для тех, чьи козы являются скорее домашними животными, чем инвестициями в сельское хозяйство.

От болезни Джона нет лекарства, и в Соединенных Штатах нет одобренной вакцины для коз, которая бы защищала их от инфекции. Следовательно, профилактика — это ключ к контролю.
Вопрос: Как мне узнать, есть ли у моего стада болезнь Джона?
A: Коза, которая выглядит совершенно здоровой, может быть инфицирована MAP. Хотя козы заражаются в первые несколько месяцев жизни, многие из них остаются свободными от клинических заболеваний до месяцев или лет.Когда козы, наконец, заболевают, симптомы расплывчаты и похожи на другие заболевания: быстрая потеря веса и, в некоторых случаях, диарея. Несмотря на то, что зараженные козы продолжают хорошо питаться, вскоре они истощаются и становятся слабыми.
Поскольку признаки болезни Джона аналогичны признакам некоторых других заболеваний — паразитизма, стоматологической болезни, казеозного лимфаденита (CLA) и вируса артрита-энцефалита коз (CAEV) — для подтверждения диагноза необходимы лабораторные тесты.
Когда обнаруживается животное с признаками болезни Джона, очень вероятно, что в стаде находятся другие инфицированные животные — даже те, которые все еще кажутся здоровыми.Борьба с инфекцией требует, чтобы вы и ваш ветеринар обращались с ней во всем стаде, а не только на отдельных животных.
Вопрос: Почему козы с клиническими признаками болезни Джона теряют вес и становятся слабыми?
A: Когда животное заражено MAP, бактерии обитают в последней части тонкой кишки — подвздошной кишке — и кишечных лимфатических узлах. В какой-то момент инфекция прогрессирует, поскольку бактерии размножаются и захватывают все больше и больше тканей. Иммунная система козы реагирует на бактерии воспалением, которое утолщает стенку кишечника и препятствует усвоению питательных веществ.В результате коза на клинической стадии болезни Джона фактически умирает от голода. На этом этапе организм также может распространяться за пределы желудочно-кишечного тракта, попадая с кровью в мышцы или другие основные органы, такие как печень или легкие.
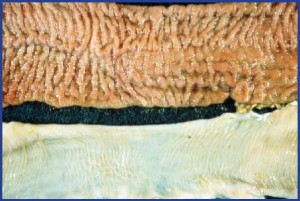
Вверху: Утолщение слизистой оболочки кишечника, вызванное болезнью Джона.
Низ: Тонкий, гибкий, нормальный кишечник
Q: Как заражаются козы? Как MAP распространяется в стаде?
A: Болезнь Джона обычно проникает в стадо при покупке зараженной, но здоровой на вид козы.С MAP, скрывающимся в тонком кишечнике, эта инфицированная коза сбрасывает организм в гранулах и в окружающую среду — возможно, на пастбище или в воду, которой пользуются ее новые сородичи. Козы подвергаются риску, когда они неоднократно заглатывают организм, особенно в молодом возрасте (менее 6 месяцев). Если самка инфицирована, ее потомство может заразиться еще до рождения (внутриутробная передача). Организм также выделяется с молоком и молозивом инфицированной оленины.
Дети наиболее восприимчивы к заражению MAP и часто заражаются при проглатывании навоза, содержащего MAP, например, при кормлении сосков, окрашенных навозом, при проглатывании молока, содержащего MAP, или поедании корма, травы или воды, содержащей навоз, загрязненный MAP.Дети, находящиеся на искусственном вскармливании, также могут заразиться, если молоко было заражено. Тепловая обработка, используемая для контроля CAE в молоке, недостаточна для уничтожения организмов MAP.
Поскольку козы обычно рожают более одного козлята за роды, болезнь Джона может быстро распространяться по стаду, особенно если инфекция остается невыявленной в течение нескольких сезонов. Хотя кажется, что существует возрастная резистентность к болезни Джона, некоторые старые козы могут заразиться, особенно когда их иммунная система подавлена по другим причинам.Болезнь Джона может передаваться от одного вида жвачных к другому, например от коров к козам, коз к овцам и т. Д.

В: Когда инфицированные животные выделяют бактерии?
A: Зараженные козы выделяют MAP в своем навозе на протяжении всей своей жизни. Чем старше животное, тем больше вероятность того, что по мере прогрессирования инфекции произойдет выделение. По мере того, как козы переходят в последнюю стадию инфекции и начинают появляться клинические признаки, инфекционный организм выделяется все чаще и тяжелее.
В: Сложно узнать, болеет ли мое стадо болезнью Джона?
A: Иногда. Болезнь Джона часто ошибочно принимают за кишечные паразиты, хроническое недоедание, токсины окружающей среды, рак и CLA, особенно у коз, у которых есть внутренние абсцессы. Многие стада меняют лечение паразитов в течение нескольких раундов, прежде чем тестировать на болезнь Джона и определять, почему их козы такие худые. Кроме того, некоторые из обычных лабораторных тестов на болезнь Джона может быть трудно интерпретировать.
Если есть подозрение на болезнь Джона, но не было подтверждено в стаде, вскрытие козы с симптомами болезни может помочь определить, есть ли инфекция в стаде. Это вскрытие может выявить увеличенные лимфатические узлы кишечника и утолщенный гофрированный кишечник.
Полное вскрытие трупа козы с подозрением на болезнь Джона должно включать посев кишечника и прилегающих лимфатических узлов, а также микроскопическое исследование этих тканей, чтобы обеспечить максимальную уверенность в диагнозе.

В: Как я могу предотвратить заражение моего стада болезнью Джона?
A: Покупатели, будьте осторожны! Наиболее распространенный способ заражения стада — покупка животного из зараженного стада. Поскольку многие люди, разводящие коз, не знают о болезни Джона, и продавец, и покупатель обычно бывают шокированы, когда ставится диагноз.
Короче говоря, легче не допустить попадания MAP в стадо, чем контролировать болезнь, когда MAP проникает внутрь.
Методы, которые могут помочь предотвратить занесение болезни Джона в стадо:
- Содержите закрытое стадо. Не купитесь на болезнь Джона.
- Если вы вводите животных в стадо, покупайте животных только из тех стад, которые прошли тестирование на болезнь Джона. В идеале приобретайте только те стада, у которых в прошлом году был отрицательный результат теста. Если это невозможно, лучше покупать у того, кто знает об инфекции, прошел тестирование на нее и может предоставить точные записи о болезни в своем стаде, чем покупать животное у того, кто никогда не слышал о болезни Джона.
- Если в исходном стаде не проводилось диагностическое тестирование, по крайней мере внимательно оцените состояние тела всех взрослых животных, обсудите историю клинических признаков у любых животных в стаде за последние несколько лет с продавцом и проверьте взрослых животных. животное для покупки.
- Если покупаемому животному меньше года, проверьте его мать, так как инфицированные молодые животные вряд ли будут иметь положительный результат на инфекцию.
- Избегайте выпаса коз на пастбищах, где паслись MAP-инфицированные жвачные животные.Выпасайте молодых коз на таком пастбище только после того, как оно год отдохнет. На сегодняшний день MAP-инфекция свободно выгуливаемых жвачных животных, таких как олени или лоси, встречается редко, и в настоящее время эти виды не считаются важным источником инфекции для сельскохозяйственных жвачных.
- Не ввозите другие виды, восприимчивые к болезни Джона: овцы, крупный рогатый скот, другие жвачные животные.
- Не садитесь на борт и не занимайте чужих коз, так как это может занести инфекцию в ваше стадо.

В: Как я могу контролировать болезнь Джона, когда она попала в мое козье стадо?
A: Поскольку болезнь Джона неизлечима, борьба с инфекцией имеет решающее значение.Борьба с болезнью Джона требует времени и твердой приверженности методам управления, направленным на то, чтобы держать молодых животных подальше от зараженного навоза, молока, кормов и воды. Типичная программа очистки стада может занять несколько лет. Основы борьбы просты: необходимо предотвращать новые инфекции, а инфицированных животных выявлять и удалять из стада.
Назначение вашего штата Координатор Johne’s может помочь вам провести оценку рисков на ферме, которая позволит оценить вашу деятельность, ваши ресурсы и ваши цели.Оценка рисков на ферме подчеркивает текущие методы управления, которые могут подвергнуть ваше стадо риску распространения болезни Джона и других инфекций. По завершении оценки риска ваш ветеринар может вместе с вами разработать план управления, разработанный специально для вас и вашего стада, который минимизирует выявленные риски передачи заболеваний. (Оценка риска на ферме является частью курса лечения болезни Джонса для производителей коз по адресу www.vetmedce.org. )
Большинство планов контроля следуют основным правилам санитарии, чтобы предотвратить передачу инфекции внутри стада.Рекомендации руководства включают:
- Не допускайте попадания навоза в места для шуток. Используйте глубокую свежую подстилку или солнечные пастбища с минимальным количеством навоза.
- Если возможно, очистите вымя маток перед кормлением грудью.
- Используйте молоко и молозиво животных с отрицательным результатом теста.
- Имейте в виду, что молозиво, приобретенное из другого козьего или коровьего стада, может быть заражено. Пастеризация должна происходить при 145 ° F (63 ° C) в течение 30 минут для периодической пастеризации или при 162 ° F (72 ° C) в течение 15 секунд для быстрой пастеризации, чтобы убить MAP в молоке.
- Подозреваемый ребенок или делает положительный тест в области, отличной от отрицательных результатов теста.
- После шуток выгоните молодняк и их матерей на «чистые» пастбища как можно скорее.
- Раннее отлучение от груди и размещение молодых коз на незагрязненных пастбищах.
- Содержите источники воды в чистоте, особенно те, которыми пользуются дети. Используйте поилки, предназначенные для минимизации фекального загрязнения.
- Поднимите все кормушки и не кормите землю.
- Используйте диагностические тесты для выявления инфицированных животных и удаления их из стада.
- Вскрытие больных или выбраковка животных, чтобы определить, заражено ли ваше стадо MAP.
- Если в вашем стаде было много случаев болезни Джона, обсудите сокращение популяции со своим ветеринаром или, как минимум, немедленно удалите всех животных с положительным тестом и их последнего ребенка. Не позволяйте детям контактировать с молоком или навозом инфицированных животных.
Помните: Профилактика болезни Джона намного дешевле, чем борьба с ней.
Q: Как я могу очистить зараженное оборудование, стойла и поля?
A: Организм MAP очень устойчив к окружающей среде: он устойчив к жаре, холоду, сушке и сырости.Хотя большинство организмов умирает через несколько месяцев, некоторые остаются в живых в течение многих месяцев. Фактически, исследования показывают, что MAP может выживать — при низких уровнях — до 11 месяцев в почве и 17 месяцев в воде. MAP также был получен из трав, удобренных навозом, загрязненным MAP. Вот почему пастбища и поля, о которых известно, что они загрязнены MAP, не должны использоваться для содержания козлят, телят или ягнят в течение как минимум одного года после последнего заражения.
Оборудование для кормления и поения, которое могло быть заражено, следует мыть и ополаскивать.При очистке поилки для воды осадок и слизь с боков и со дна не следует сбрасывать на землю, где будут пастись молодые козы, так как осадок и слизь могут быть загрязнены MAP.
Дезинфицирующие средства, помеченные как «туберкулоцидные», можно использовать по назначению для очистки инструментов, инвентаря и некоторых поверхностей. Эти дезинфицирующие средства, однако, могут быть инактивированы органическими материалами, такими как грязь и навоз, и поэтому неэффективны для грязных поверхностей, деревянных поверхностей, почвы или даже цементных полов.
Компостирование навоза и использованных подстилок может снизить количество живых организмов MAP.

В: Следует ли мне проверить свое стадо на болезнь Джона?
A: Если у вас есть козы с нормальным аппетитом, которые похудели и не реагируют на лечение, поговорите со своим ветеринаром. Причиной может быть болезнь Джона.
Помните: Поскольку болезнь Джона — проблема стада, тестирование должно быть сосредоточено на стаде, а не только на одном животном.
Диагностическое тестирование на болезнь Джона может помочь:
- Определите, присутствует ли инфекция MAP в вашем стаде.
- Оцените степень заражения MAP в вашем стаде.
- Контрольный MAP в зараженном стаде.
- Поставьте диагноз больному животному.
- Проверьте, присутствует ли MAP в окружающей среде.
- Выполните условия предварительной покупки или доставки.
- Продемонстрируйте потенциальным покупателям, что у ваших животных низкий риск болезни Джона (отрицательный результат теста).Как только ваш ветеринар узнает ваши цели в тестировании на болезнь Джона, можно разработать план тестирования, который наилучшим образом соответствует вашим потребностям. В этом плане следует указать тип теста, когда проводить тестирование, на каких животных следует сосредоточить внимание, стоимость тестирования, как интерпретировать результаты и какие действия следует предпринять на основе результатов тестирования. Решите, как вы планируете использовать результаты ваших тестов, прежде чем собирать образцы.
Q: Какие диагностические тесты доступны? Какой лучше?
A: Несмотря на то, что не существует единого «лучшего теста» на болезнь Джона у коз, лучший план тестирования — это план, разработанный вами и вашим ветеринаром, поскольку вы лучше всего знаете свое дело — его цели, ресурсы и другие проблемы со здоровьем животных.
Диагностические тесты на болезнь Джона ищут либо организм, вызывающий болезнь Джона (MAP), либо реакцию животного на инфекцию.
Тесты для поиска микроорганизмов включают посев и ПЦР с тестированием образцов навоза. Можно протестировать отдельных животных, или лаборатория может объединить образцы навоза от нескольких животных и предоставить владельцам эффективный надзор за болезнью Джона за небольшую часть стоимости индивидуального посева или ПЦР.
Организм животного реагирует на инфекцию, вырабатывая антитела.Тесты, измеряющие уровни антител, — это ELISA для образцов молока и крови. Из-за биологии MAP-инфекции у коз более старшего возраста вероятность выделения MAP или выработки антител гораздо выше. Поэтому диагностические тесты менее надежны для коз младше 18 месяцев.

Подходы к тестированию, которые хорошо зарекомендовали себя на других стадах коз, включают:
| Цель тестирования | Вариант A | Вариант B |
|---|---|---|
| Подтвердите наличие MAP в стаде. | Культура 5 — 10 проб фекалий окружающей среды, взятых на ферме в местах с интенсивным движением коз. | Используя ELISA * или культуру фекалий, проверьте самых старых или худых коз — 10% или более от стада. |
| Определите количество зараженных коз. | Анализ крови (ELISA *) всех взрослых коз. | Соберите образцы фекалий для тестирования в лаборатории путем объединения для культивирования. Для каждого положительного пула образцы повторно тестируются индивидуально. |
| Контроль или искоренение MAP в инфицированном стаде. | Анализ крови (ELISA *) коз после второго шучу или старше. | Соберите образцы фекалий для тестирования в лаборатории путем объединения для культивирования. Для каждого положительного пула образцы повторно тестируются индивидуально. |
| Диагностируйте заболевание козы или козы на потерю веса и диарею. | Если предыдущие случаи были зарегистрированы в стаде: ELISA. * (Посев фекалий, если CLA является проблемой в стаде или если стадо было вакцинировано от CLA.) | Если MAP никогда не подтверждался в стаде, используйте фекальный посев.* Используйте коммерческий набор ELISA, одобренный Министерством сельского хозяйства США для мелких жвачных, чтобы ограничить вероятность ложноположительных результатов из-за перекрестной реакции антител от других типов инфекций. Образцы для испытаний следует отправлять в лабораторию, которая прошла ежегодный «контрольный тест», подтверждающий их компетентность. Эти лаборатории перечислены здесь. |
Q: Где я могу найти дополнительную информацию о болезни Джона?
A: Веб-сайт Школы ветеринарной медицины Университета Висконсина по болезни Джона — www.johnes.org — затрагивает все аспекты болезни Джона у различных видов животных, включая коз. На сайте есть функция «Задайте вопрос эксперту», которая позволяет вам задавать свои вопросы и получать индивидуальный ответ от эксперта.
Школа ветеринарной медицины Университета Висконсина также предлагает бесплатный онлайн-курс для производителей коз. Просто зайдите на сайт www.vetmedce.org и нажмите «Курсы» в нижнем левом углу главной страницы. Оказавшись на новой странице, нажмите «Болезнь Джона». На следующей новой странице нажмите «Курсы по болезни Джона для производителей», а затем нажмите «0017 — Болезнь Джона для производителей коз».”
Чтобы узнать больше о болезни Джона у коз, обратитесь в ваше государственное агентство по контролю за здоровьем животных или к координатору Johne’s, назначенному вашим штатом. Контактная информация программы по болезни Джона в вашем штате доступна на сайте www.johnesdisease.org, когда вы нажимаете «State Contacts».


ВОЗ | Всемирная организация здравоохранения
Забытые тропические болезни (NTD) — разнообразная группа инфекционных болезней, которые преобладают в тропических и субтропических условиях в 149 странах — поражают более одного миллиарда человек и ежегодно обходятся развивающимся странам в миллиарды долларов. Больше всего страдают люди, живущие в бедности, без надлежащей санитарии и в тесном контакте с переносчиками инфекций, домашними животными и домашним скотом.
Эффективный контроль может быть достигнут, если отдельные подходы общественного здравоохранения объединены и реализованы на местном уровне.Вмешательства регулируются местной эпидемиологией и наличием соответствующих мер для обнаружения, предотвращения и контроля заболеваний. Осуществление соответствующих мер с высоким уровнем охвата будет способствовать достижению целей Дорожной карты ВОЗ по борьбе с заболеванием и лечением забытых тропических болезней, в результате чего к 2020 году будут устранены многие и по крайней мере два.
В мае 2013 г. 66-я сессия Всемирной ассамблеи здравоохранения постановила активизировать и интегрировать меры по борьбе с забытыми тропическими болезнями и запланировать инвестиции для улучшения здоровья и социального благополучия затронутого населения.ВОЗ работает с государствами-членами для обеспечения выполнения резолюции WHA66.12.
В 2016 г. 69-я Ассамблея приняла резолюцию WHA69.21 о бремени мицетомы и обратилась к ВОЗ с просьбой через Стратегические и технические консультации по забытым тропическим болезням «определить систематический, технически управляемый процесс оценки и возможное включение дополнительных болезни среди «забытых тропических болезней» ».
Соответственно, в 2017 г. на 10-м совещании Стратегической и технической консультативной группы по забытым тропическим болезням были получены предложения о добавлении болезней и, в соответствии с необходимыми процедурами, хромобластомикоза и других глубоких микозов , чесотки и других эктопаразитов и Змеиный яд был добавлен в портфель NTD:
.